Elegans: Read Alignment and Count Table with QausR and Rhisat2
Lieven Clement
statOmics, Ghent University (https://statomics.github.io)
1 Background
After fertilization but prior to the onset of zygotic transcription, the C. elegans zygote cleaves asymmetrically to create the anterior AB and posterior P1 blastomeres, each of which goes on to generate distinct cell lineages. To understand how patterns of RNA inheritance and abundance arise after this first asymmetric cell division, we pooled hand-dissected AB and P1 blastomeres and performed RNA-seq (Study GSE59943).
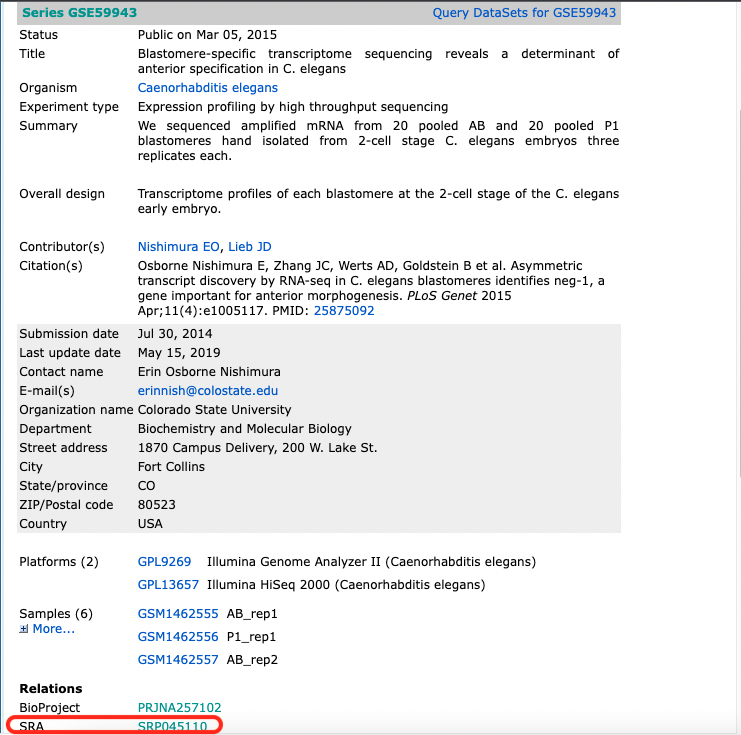
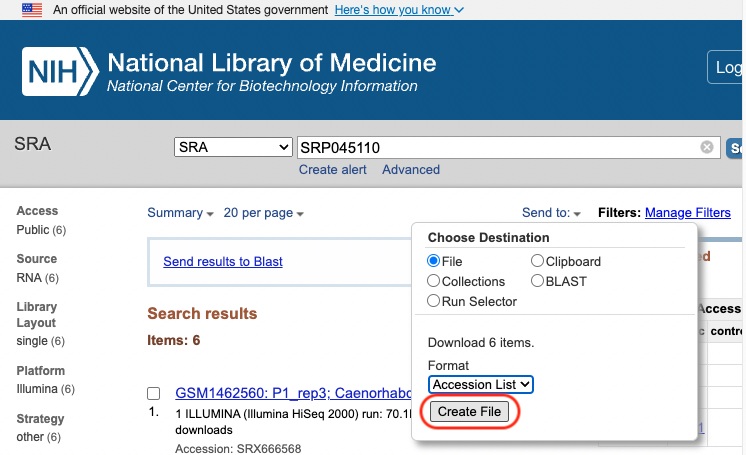
The information which connects the sample information from GEO with the SRA run id is downloaded from SRA using the Send to: File button.
2 Preprocessing
2.1 Download Data
We used the info in the downloaded SraAccList.txt file to download the SRA files. We used the system function to invoke aan OS command. xargs can converts each line of a file into an argument.
The fasterq-dump function will then download and convert each corresponding sra file from the SRA archive to a fastq file.
The fasterq-dump is a tool from the sra-tools suite that can be used to download sra files and to convert them into the fastq format.
system("xargs fasterq-dump -p < SraAccList.txt")For illustration purposes we will download subsampled fastq files from the course github site.
timeout <- options()$timeout
options(timeout = 300) #prevent timeout
url <- "https://github.com/statOmics/SGA/archive/refs/heads/elegansFastq.zip"
destFile <- "elegansFastq.zip"
download.file(url, destFile)
unzip(destFile, exdir = "./", overwrite = TRUE)
if (file.exists(destFile)) {
#Delete file if it exists
file.remove(destFile)
}## [1] TRUE2.2 Assess Read Quality
Assess the fastq files with fastQC.
2.3 Align data
2.3.1 Input file for hisat aligner
We will make the input file for the hisat aligner that will be called through the QUASAR package.
fastqFiles <- list.files(path = "SGA-elegansFastq",full.names = TRUE, pattern = "fastq")
samples <- fastqFiles |>
substr(start=18,stop=27)
fileInfo <- tibble(FileName=fastqFiles,SampleName=samples)
fileInfoWe write the fileInfo to disk
2.3.2 Download C. elegans genome and gff3 file

slide courtesy Charlotte Soneson
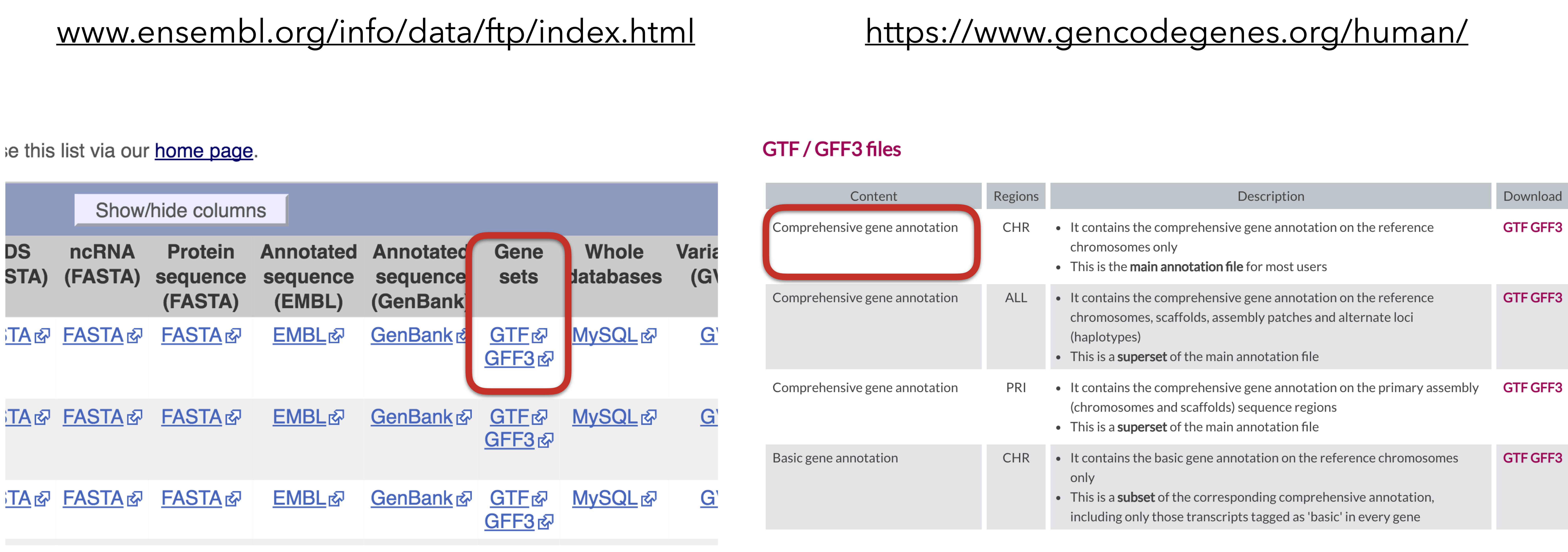
slide courtesy Charlotte Soneson
urlGenome <- "https://ftp.ensembl.org/pub/release-108/fasta/caenorhabditis_elegans/dna/Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.gz"
genomeDestFile <- "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.gz"
urlGff <- "https://ftp.ensembl.org/pub/release-108/gff3/caenorhabditis_elegans/Caenorhabditis_elegans.WBcel235.108.gff3.gz"
gffDestFile <- "Caenorhabditis_elegans.WBcel235.108.gff3.gz"
download.file(url = urlGenome ,
destfile = genomeDestFile)
download.file(url = urlGff,
destfile = gffDestFile)
gunzip(genomeDestFile, overwrite = TRUE, remove=TRUE)
gunzip(gffDestFile, overwrite = TRUE, remove=TRUE)
list.files()## [1] "_session-info.Rmd"
## [2] "_site.yml"
## [3] "about.html"
## [4] "about.Rmd"
## [5] "airway_files"
## [6] "airway_salmon_edgeR_files"
## [7] "airway_salmon_edgeR.html"
## [8] "airway_salmon_edgeR.Rmd"
## [9] "airway.html"
## [10] "airway.rmd"
## [11] "airwayCountTable.csv"
## [12] "airwayMapping.html"
## [13] "airwayMapping.Rmd"
## [14] "bibliography.bib"
## [15] "Caenorhabditis_elegans.WBcel235.108.gff3"
## [16] "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa"
## [17] "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.fai"
## [18] "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.md5"
## [19] "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa.Rhisat2"
## [20] "cancer3x3_files"
## [21] "cancer3x3.html"
## [22] "cancer3x3.pdf"
## [23] "cancer3x3.Rmd"
## [24] "cancer6x6_files"
## [25] "cancer6x6.html"
## [26] "cancer6x6.pdf"
## [27] "cancer6x6.Rmd"
## [28] "cancer9x9_files"
## [29] "cancer9x9.html"
## [30] "cancer9x9.pdf"
## [31] "cancer9x9.Rmd"
## [32] "cptac_maxLFQ_files"
## [33] "cptac_maxLFQ.html"
## [34] "cptac_maxLFQ.pdf"
## [35] "cptac_maxLFQ.Rmd"
## [36] "cptac_median_files"
## [37] "cptac_median.html"
## [38] "cptac_median.pdf"
## [39] "cptac_median.Rmd"
## [40] "cptac_robust_files"
## [41] "cptac_robust_gui.html"
## [42] "cptac_robust_gui.Rmd"
## [43] "cptac_robust.html"
## [44] "cptac_robust.pdf"
## [45] "cptac_robust.Rmd"
## [46] "docs"
## [47] "elegansAlignmentCountTable.Rmd"
## [48] "elegansCountTable.csv"
## [49] "elegansDE.Rmd"
## [50] "fastQfiles.txt"
## [51] "figs"
## [52] "figures"
## [53] "figures.tar.gz"
## [54] "footer.html"
## [55] "heartMainInteraction.Rmd"
## [56] "Homo_sapiens.GRCh38.113.chromosome.1.gff3"
## [57] "Homo_sapiens.GRCh38.dna.chromosome.1.fa"
## [58] "Homo_sapiens.GRCh38.dna.chromosome.1.fa.fai"
## [59] "Homo_sapiens.GRCh38.dna.chromosome.1.fa.md5"
## [60] "Homo_sapiens.GRCh38.dna.chromosome.1.fa.Rhisat2"
## [61] "images_sequencing"
## [62] "index.Rmd"
## [63] "install.R"
## [64] "intro.Rmd"
## [65] "msqrob2.bib"
## [66] "multipleRegression_KPNA2_files"
## [67] "multipleRegression_KPNA2.Rmd"
## [68] "mzIDMsgfSwissprotExample.Rmd"
## [69] "mzIDswissprotTutorial.Rmd"
## [70] "mzIDuniprotTutorial.Rmd"
## [71] "parathyroid_files"
## [72] "parathyroid.Rmd"
## [73] "pda_blocking_wrapup_files"
## [74] "pda_blocking_wrapup.Rmd"
## [75] "pda_quantification_inference_files"
## [76] "pda_quantification_inference.Rmd"
## [77] "pda_quantification_preprocessing_files"
## [78] "pda_quantification_preprocessing.Rmd"
## [79] "pda_robustSummarisation_peptideModels_files"
## [80] "pda_robustSummarisation_peptideModels.Rmd"
## [81] "pda_technicalDetails.Rmd"
## [82] "pda_tutorialDesign.Rmd"
## [83] "pda_tutorialPreprocessing.Rmd"
## [84] "pyrococcusMSGF+.mzid"
## [85] "pyroSwissprot.mzid"
## [86] "pyroUniprot.mzid"
## [87] "QuasR_log_debd2f67fea0.txt"
## [88] "QuasR_log_e34943403f50.txt"
## [89] "README.md"
## [90] "recapGeneralLinearModel_files"
## [91] "recapGeneralLinearModel.Rmd"
## [92] "rmdBub"
## [93] "scRNA-seq-Tutorial.Rmd"
## [94] "sequencing_countData_files"
## [95] "sequencing_countData.log"
## [96] "sequencing_countData.Rmd"
## [97] "sequencing_intro.log"
## [98] "sequencing_intro.Rmd"
## [99] "sequencing_singleCell_files"
## [100] "sequencing_singleCell.Rmd"
## [101] "sequencing_technicalDE_files"
## [102] "sequencing_technicalDE.Rmd"
## [103] "sequencingTutorial1_mapping.Rmd"
## [104] "sequencingTutorial2_DE.Rmd"
## [105] "sequencingTutorial3_DE.Rmd"
## [106] "SGA-airwaySeqData"
## [107] "SGA-airwaySeqData.zip"
## [108] "SGA-elegansFastq"
## [109] "singleCell_MacoskoWorkflow_blank.Rmd"
## [110] "site_libs"
## [111] "style.css"
## [112] "TargetDecoy.Rmd"
## [113] "TargetDecoyTutorial.Rmd"2.3.3 Align reads using QuasR and RHisat package
We are aligning RNA-seq reads and have to use a gap aware alignment modus. We therefore use the argument
splicedAlignment = TRUEThe project is a single-end sequencing project! So we use the argument
paired = "no".
suppressPackageStartupMessages({
library(QuasR)
library(Rhisat2)
})
sampleFile <- "fastQfiles.txt"
genomeFile <- "Caenorhabditis_elegans.WBcel235.dna.toplevel.fa"
projElegans <- qAlign(
sampleFile = sampleFile,
genome = genomeFile,
splicedAlignment = TRUE,
aligner = "Rhisat2",
paired="no")## all necessary alignment files found2.4 Make count table
2.4.1 Convert gff file into database
The gff3 file contains information on the position of features, e.g. exons and genes, along the chromosome.
suppressPackageStartupMessages({
library(GenomicFeatures)
library(Rsamtools)
})
annotFile <- "Caenorhabditis_elegans.WBcel235.108.gff3"
#chrLen <- scanFaIndex(genomeFile)
#chrominfo <- data.frame(chrom = as.character(seqnames(chrLen)),
# length = width(chrLen),
# is_circular = rep(FALSE, length(chrLen)))
txdb <- makeTxDbFromGFF(file = annotFile,
dataSource = "Ensembl",
organism = "Caenorhabditis elegans")## Warning in call_fun_in_txdbmaker("makeTxDbFromGFF", ...): makeTxDbFromGFF() has moved to the txdbmaker package. Please call
## txdbmaker::makeTxDbFromGFF() to get rid of this warning.## Import genomic features from the file as a GRanges object ... OK
## Prepare the 'metadata' data frame ... OK
## Make the TxDb object ...## Warning in .extract_exons_from_GRanges(exon_IDX, gr, mcols0, tx_IDX, feature = "exon", : 15639 exons couldn't be linked to a transcript so were dropped (showing
## only the first 6):
## seqid start end strand ID Name Parent Parent_type
## 1 I 31523 31543 + <NA> Y74C9A.7.e1 transcript:Y74C9A.7 <NA>
## 2 I 32415 32435 + <NA> Y74C9A.8.e1 transcript:Y74C9A.8 <NA>
## 3 I 115656 115676 - <NA> F53G12.14.e1 transcript:F53G12.14 <NA>
## 4 I 272603 272623 - <NA> C53D5.8.e1 transcript:C53D5.8 <NA>
## 5 I 272607 272627 - <NA> C53D5.7.e1 transcript:C53D5.7 <NA>
## 6 I 463304 463324 + <NA> W04C9.7.e1 transcript:W04C9.7 <NA>## OK2.4.2 Make Gene Count table
With the qCount function we can count the overlap between the aligned reads and the genomic features of interest.
## extracting gene regions from TxDb...done
## counting alignments...done
## collapsing counts by query name...done## width SRR1532959 SRR1532960 SRR1532961 SRR1532962 SRR1532963
## WBGene00000001 1806 109 57 135 67 202
## WBGene00000002 1739 0 0 0 0 1
## WBGene00000003 1738 1 3 4 4 3
## WBGene00000004 2027 3 2 2 1 3
## WBGene00000005 1821 0 0 0 0 0
## WBGene00000006 1937 0 0 0 0 0
## SRR1532964
## WBGene00000001 147
## WBGene00000002 1
## WBGene00000003 6
## WBGene00000004 5
## WBGene00000005 0
## WBGene00000006 0Save count table for future use.
3 Session Info
With respect to reproducibility, it is highly recommended to include a session info in your script so that readers of your output can see your particular setup of R.
## R version 4.4.0 RC (2024-04-16 r86468)
## Platform: aarch64-apple-darwin20
## Running under: macOS Big Sur 11.6
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: Europe/Brussels
## tzcode source: internal
##
## attached base packages:
## [1] stats4 parallel stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] Rsamtools_2.20.0 Biostrings_2.72.1 XVector_0.44.0
## [4] GenomicFeatures_1.56.0 AnnotationDbi_1.66.0 Biobase_2.64.0
## [7] Rhisat2_1.20.0 QuasR_1.44.0 Rbowtie_1.44.0
## [10] GenomicRanges_1.56.1 GenomeInfoDb_1.40.1 IRanges_2.38.1
## [13] S4Vectors_0.42.1 BiocGenerics_0.50.0 R.utils_2.12.3
## [16] R.oo_1.26.0 R.methodsS3_1.8.2 lubridate_1.9.3
## [19] forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4
## [22] purrr_1.0.2 readr_2.1.5 tidyr_1.3.1
## [25] tibble_3.2.1 ggplot2_3.5.1 tidyverse_2.0.0
##
## loaded via a namespace (and not attached):
## [1] DBI_1.2.3 bitops_1.0-8
## [3] deldir_2.0-4 httr2_1.0.5
## [5] biomaRt_2.60.1 rlang_1.1.4
## [7] magrittr_2.0.3 matrixStats_1.4.1
## [9] compiler_4.4.0 RSQLite_2.3.7
## [11] txdbmaker_1.0.1 png_0.1-8
## [13] vctrs_0.6.5 pwalign_1.0.0
## [15] pkgconfig_2.0.3 crayon_1.5.3
## [17] fastmap_1.2.0 dbplyr_2.5.0
## [19] utf8_1.2.4 rmarkdown_2.28
## [21] tzdb_0.4.0 UCSC.utils_1.0.0
## [23] bit_4.5.0 xfun_0.47
## [25] zlibbioc_1.50.0 cachem_1.1.0
## [27] jsonlite_1.8.9 progress_1.2.3
## [29] blob_1.2.4 highr_0.11
## [31] DelayedArray_0.30.1 BiocParallel_1.38.0
## [33] jpeg_0.1-10 prettyunits_1.2.0
## [35] R6_2.5.1 VariantAnnotation_1.50.0
## [37] bslib_0.8.0 stringi_1.8.4
## [39] RColorBrewer_1.1-3 rtracklayer_1.64.0
## [41] jquerylib_0.1.4 Rcpp_1.0.13-1
## [43] SummarizedExperiment_1.34.0 knitr_1.48
## [45] SGSeq_1.38.0 igraph_2.0.3
## [47] Matrix_1.7-0 timechange_0.3.0
## [49] tidyselect_1.2.1 rstudioapi_0.16.0
## [51] abind_1.4-8 yaml_2.3.10
## [53] codetools_0.2-20 RUnit_0.4.33
## [55] hwriter_1.3.2.1 curl_5.2.3
## [57] lattice_0.22-6 withr_3.0.1
## [59] ShortRead_1.62.0 KEGGREST_1.44.1
## [61] evaluate_1.0.0 BiocFileCache_2.12.0
## [63] xml2_1.3.6 filelock_1.0.3
## [65] pillar_1.9.0 MatrixGenerics_1.16.0
## [67] generics_0.1.3 vroom_1.6.5
## [69] RCurl_1.98-1.16 hms_1.1.3
## [71] munsell_0.5.1 scales_1.3.0
## [73] GenomicFiles_1.40.0 glue_1.8.0
## [75] tools_4.4.0 interp_1.1-6
## [77] BiocIO_1.14.0 BSgenome_1.72.0
## [79] GenomicAlignments_1.40.0 XML_3.99-0.17
## [81] grid_4.4.0 latticeExtra_0.6-30
## [83] colorspace_2.1-1 GenomeInfoDbData_1.2.12
## [85] restfulr_0.0.15 cli_3.6.3
## [87] rappdirs_0.3.3 fansi_1.0.6
## [89] S4Arrays_1.4.1 gtable_0.3.5
## [91] sass_0.4.9 digest_0.6.37
## [93] SparseArray_1.4.8 rjson_0.2.23
## [95] memoise_2.0.1 htmltools_0.5.8.1
## [97] lifecycle_1.0.4 httr_1.4.7
## [99] bit64_4.5.2