Proteomics data analysis: heart
Lieven Clement
statOmics, Ghent University (https://statomics.github.io)

This is part of the online course Proteomics Data Analysis (PDA)
1 Background
Researchers have assessed the proteome in different regions of the heart for 3 patients (identifiers 3, 4, and 8). For each patient they sampled the left atrium (LA), right atrium (RA), left ventricle (LV) and the right ventricle (RV). The data are a small subset of the public dataset PXD006675 on PRIDE.
Suppose that researchers are mainly interested in comparing the ventricular to the atrial proteome. Particularly, they would like to compare the left atrium to the left ventricle, the right atrium to the right ventricle, the average ventricular vs atrial proteome and if ventricular vs atrial proteome shifts differ between left and right heart region.
Representation of the heart
2 Data
We first import the peptides.txt file. This is the file that contains your peptide-level intensities. For a MaxQuant search [6], this peptides.txt file can be found by default in the “path_to_raw_files/combined/txt/” folder from the MaxQuant output, with “path_to_raw_files” the folder where raw files were saved. In this tutorial, we will use a MaxQuant peptides file from MaxQuant that can be found in the data tree of the SGA2020 github repository https://github.com/statOmics/SGA2020/tree/data/quantification/heart .
2.1 Data import
Click to see background and code
To import the data we use the QFeatures package.
We generate the object peptideRawFile with the path to the peptideRaws.txt file. Using the grepEcols function, we find the columns that contain the expression data of the peptideRaws in the peptideRaws.txt file.
library(tidyverse)
library(limma)
library(QFeatures)
library(msqrob2)
library(plotly)
peptidesFile <- "https://raw.githubusercontent.com/statOmics/PDA21/data/quantification/heart/peptides.txt"
ecols <- grep("Intensity\\.", names(read.delim(peptidesFile)))
pe <- readQFeatures(
table = peptidesFile,
fnames = 1,
ecol = ecols,
name = "peptideRaw", sep="\t")
pe## An instance of class QFeatures containing 1 assays:
## [1] peptideRaw: SummarizedExperiment with 31319 rows and 12 columnspe[["peptideRaw"]]## class: SummarizedExperiment
## dim: 31319 12
## metadata(0):
## assays(1): ''
## rownames(31319): AAAAAAAAAK AAAAAAAAEQQSSNGPVK ... YYTPVPCESATAK
## YYTYLIMNK
## rowData names(91): Sequence N.term.cleavage.window ...
## Oxidation..M..site.IDs MS.MS.Count
## colnames(12): Intensity.LA3 Intensity.LA4 ... Intensity.RV4
## Intensity.RV8
## colData names(0):We will make use from data wrangling functionalities from the tidyverse package. The %>% operator allows us to pipe the output of one function to the next function.
colData(pe)$location <- substr(
colnames(pe[["peptideRaw"]]),
11,
11) %>%
unlist %>%
as.factor
colData(pe)$tissue <- substr(
colnames(pe[["peptideRaw"]]),
12,
12) %>%
unlist %>%
as.factor
colData(pe)$patient <- substr(
colnames(pe[["peptideRaw"]]),
13,
13) %>%
unlist %>%
as.factorWe calculate how many non zero intensities we have per peptide and this will be useful for filtering.
rowData(pe[["peptideRaw"]])$nNonZero <- rowSums(assay(pe[["peptideRaw"]]) > 0)Peptides with zero intensities are missing peptides and should be represent with a NA value rather than 0.
pe <- zeroIsNA(pe, "peptideRaw") # convert 0 to NA2.2 Data exploration
Click to see background and code
63% of all peptide intensities are missing and for some peptides we do not even measure a signal in any sample. The missingness is similar across samples.
3 Preprocessing
Click to see background and code
This section preforms standard preprocessing for the peptide data. This include log transformation, filtering and summarisation of the data.
3.1 Log transform the data
pe <- logTransform(pe, base = 2, i = "peptideRaw", name = "peptideLog")
limma::plotDensities(assay(pe[["peptideLog"]]))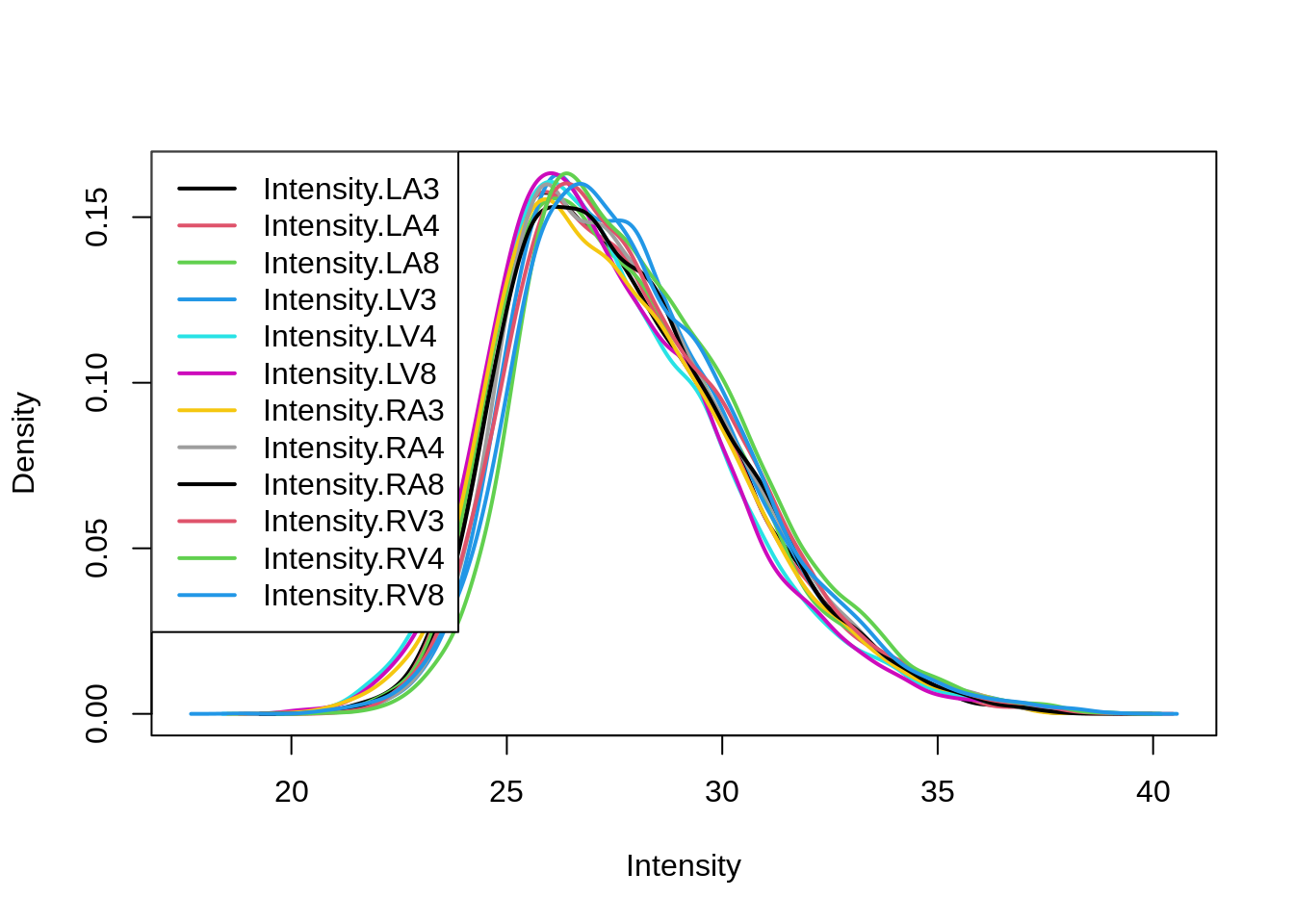
3.2 Filtering
Click to see background and code
3.2.1 Handling overlapping protein groups
In our approach a peptide can map to multiple proteins, as long as there is none of these proteins present in a smaller subgroup.
pe <- filterFeatures(pe, ~ Proteins %in% smallestUniqueGroups(rowData(pe[["peptideLog"]])$Proteins))3.2.2 Remove reverse sequences (decoys) and contaminants
We now remove the contaminants, peptides that map to decoy sequences, and proteins which were only identified by peptides with modifications.
First look to the names of the variables for the peptide features
pe[["peptideLog"]] %>%
rowData %>%
names## [1] "Sequence" "N.term.cleavage.window"
## [3] "C.term.cleavage.window" "Amino.acid.before"
## [5] "First.amino.acid" "Second.amino.acid"
## [7] "Second.last.amino.acid" "Last.amino.acid"
## [9] "Amino.acid.after" "A.Count"
## [11] "R.Count" "N.Count"
## [13] "D.Count" "C.Count"
## [15] "Q.Count" "E.Count"
## [17] "G.Count" "H.Count"
## [19] "I.Count" "L.Count"
## [21] "K.Count" "M.Count"
## [23] "F.Count" "P.Count"
## [25] "S.Count" "T.Count"
## [27] "W.Count" "Y.Count"
## [29] "V.Count" "U.Count"
## [31] "O.Count" "Length"
## [33] "Missed.cleavages" "Mass"
## [35] "Proteins" "Leading.razor.protein"
## [37] "Start.position" "End.position"
## [39] "Gene.names" "Protein.names"
## [41] "Unique..Groups." "Unique..Proteins."
## [43] "Charges" "PEP"
## [45] "Score" "Identification.type.LA3"
## [47] "Identification.type.LA4" "Identification.type.LA8"
## [49] "Identification.type.LV3" "Identification.type.LV4"
## [51] "Identification.type.LV8" "Identification.type.RA3"
## [53] "Identification.type.RA4" "Identification.type.RA8"
## [55] "Identification.type.RV3" "Identification.type.RV4"
## [57] "Identification.type.RV8" "Fraction.Average"
## [59] "Fraction.Std..Dev." "Fraction.1"
## [61] "Fraction.2" "Fraction.3"
## [63] "Fraction.4" "Fraction.5"
## [65] "Fraction.6" "Fraction.7"
## [67] "Fraction.8" "Fraction.100"
## [69] "Experiment.LA3" "Experiment.LA4"
## [71] "Experiment.LA8" "Experiment.LV3"
## [73] "Experiment.LV4" "Experiment.LV8"
## [75] "Experiment.RA3" "Experiment.RA4"
## [77] "Experiment.RA8" "Experiment.RV3"
## [79] "Experiment.RV4" "Experiment.RV8"
## [81] "Intensity" "Reverse"
## [83] "Potential.contaminant" "id"
## [85] "Protein.group.IDs" "Mod..peptide.IDs"
## [87] "Evidence.IDs" "MS.MS.IDs"
## [89] "Best.MS.MS" "Oxidation..M..site.IDs"
## [91] "MS.MS.Count" "nNonZero"No information on decoys.
pe <- filterFeatures(pe,~ Potential.contaminant != "+")3.2.3 Drop peptides that were only identified in one sample
We keep peptides that were observed at least twice.
pe <- filterFeatures(pe,~nNonZero >= 2)
nrow(pe[["peptideLog"]])## [1] 17432We keep 17432 peptides after filtering.
3.3 Normalize the data
Click to see background and code
pe <- normalize(pe,
i = "peptideLog",
name = "peptideNorm",
method = "center.median")3.3.1 Explore normalized data
Click to see background and code
After normalisation the density curves for all samples are comparable.
limma::plotDensities(assay(pe[["peptideNorm"]]))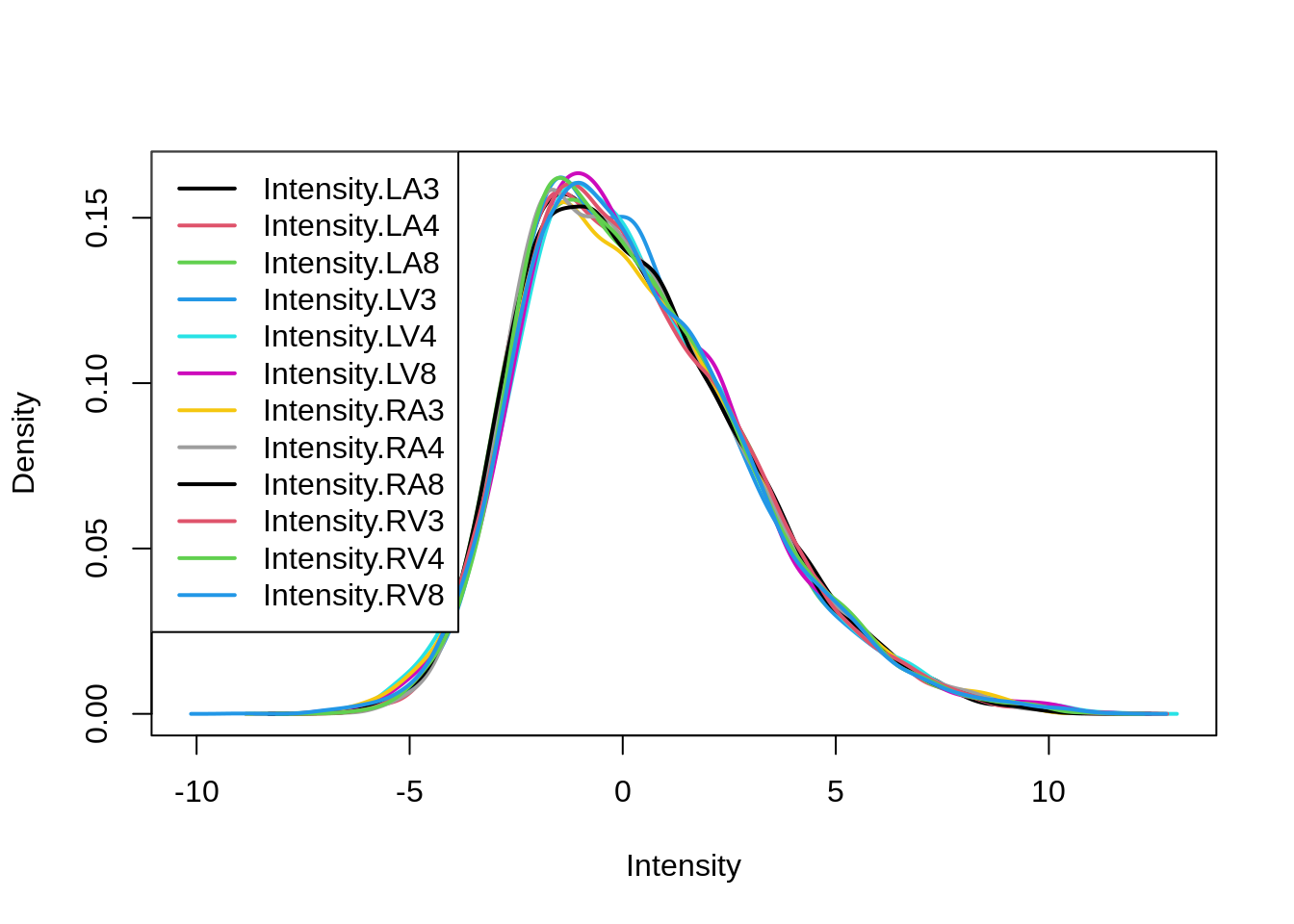
This is more clearly seen is a boxplot.
boxplot(assay(pe[["peptideNorm"]]), col = palette()[-1],
main = "Peptide distribtutions after normalisation", ylab = "intensity")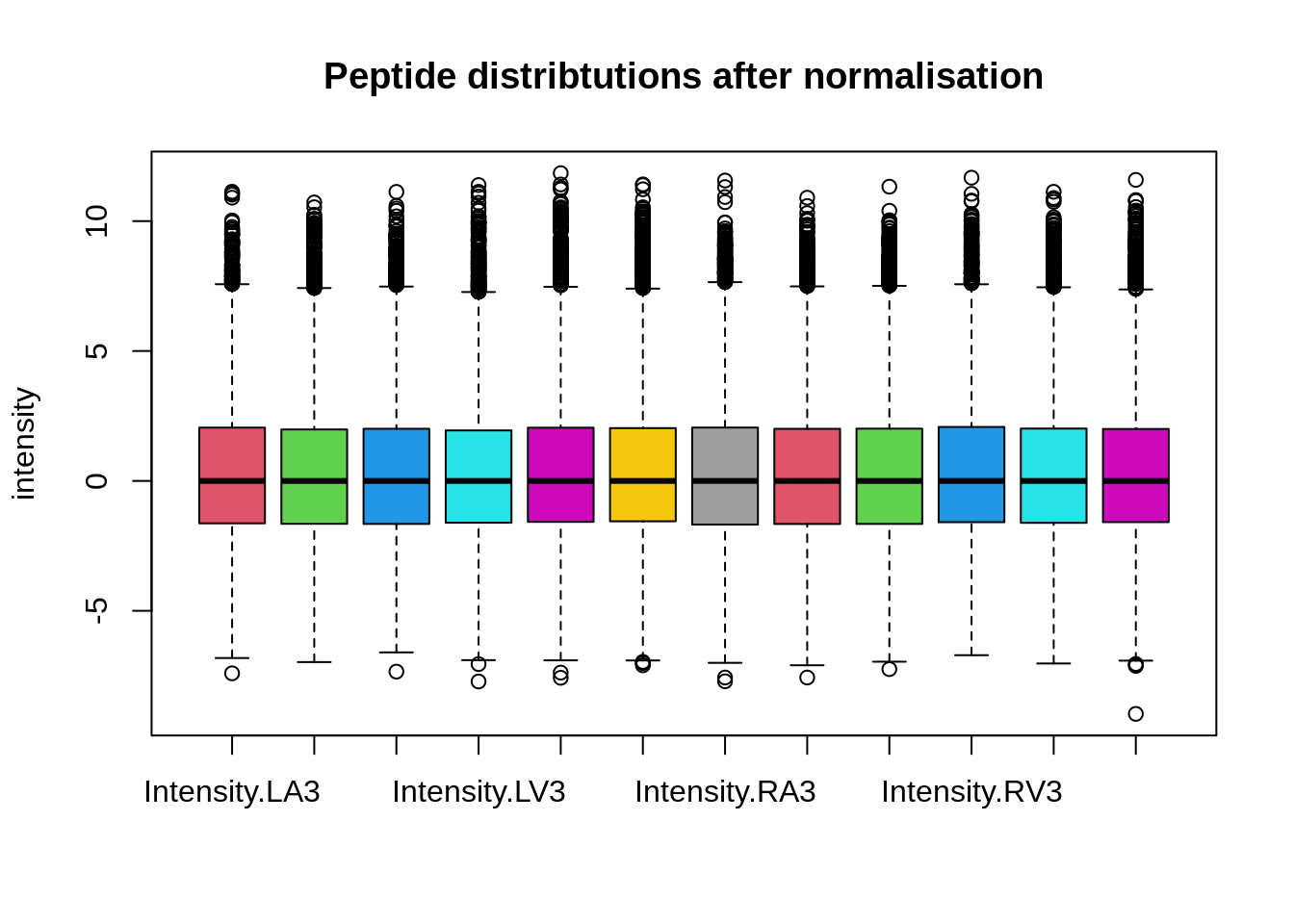
We can visualize our data using a Multi Dimensional Scaling plot, eg. as provided by the limma package.
limma::plotMDS(assay(pe[["peptideNorm"]]),
col = colData(pe)$location:colData(pe)$tissue %>%
as.numeric,
labels = colData(pe) %>%
rownames %>%
substr(start = 11, stop = 13)
)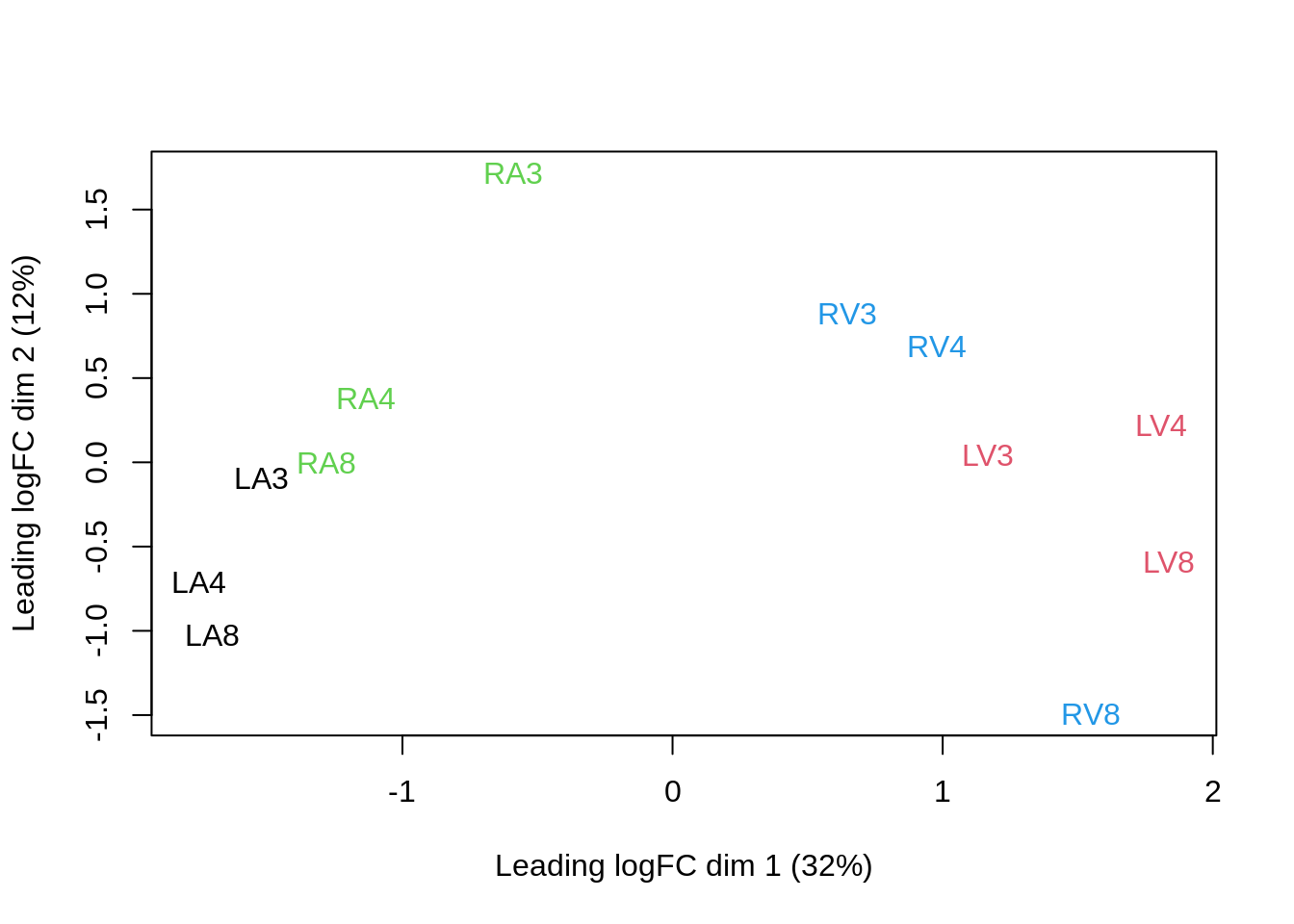
The first axis in the plot is showing the leading log fold changes (differences on the log scale) between the samples.
3.4 Summarization to protein level
Click to see background and code
We use robust summarization in aggregateFeatures. This is the default workflow of aggregateFeatures so you do not have to specifiy the argument fun. However, because we compare methods we have included the fun argument to show the summarization method explicitely.
pe <- aggregateFeatures(pe,
i = "peptideNorm",
fcol = "Proteins",
na.rm = TRUE,
name = "proteinRobust",
fun = MsCoreUtils::robustSummary)## Your quantitative and row data contain missing values. Please read the
## relevant section(s) in the aggregateFeatures manual page regarding the
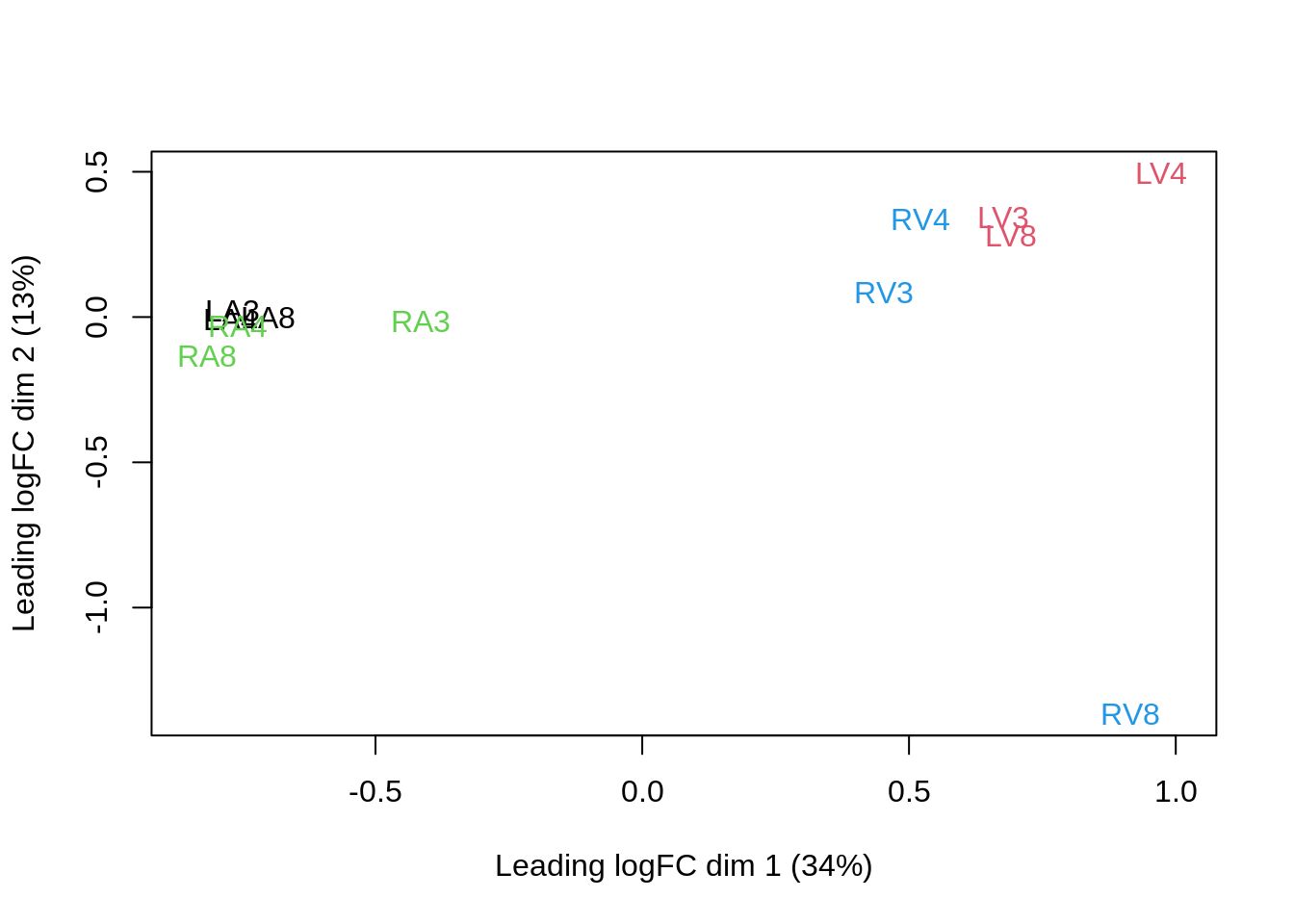
## effects of missing values on data aggregation.plotMDS(assay(pe[["proteinRobust"]]),
col = colData(pe)$location:colData(pe)$tissue %>%
as.numeric,
labels = colData(pe) %>%
rownames %>%
substr(start = 11, stop = 13)
)
4 Data Analysis
4.1 Estimation
We model the protein level expression values using msqrob. By default msqrob2 estimates the model parameters using robust regression.
pe <- msqrob(
object = pe,
i = "proteinRobust",
formula = ~ location*tissue + patient)4.2 Inference
Explore Design
library(ExploreModelMatrix)
VisualizeDesign(colData(pe),~ location*tissue + patient)$plotlist## $`location = L`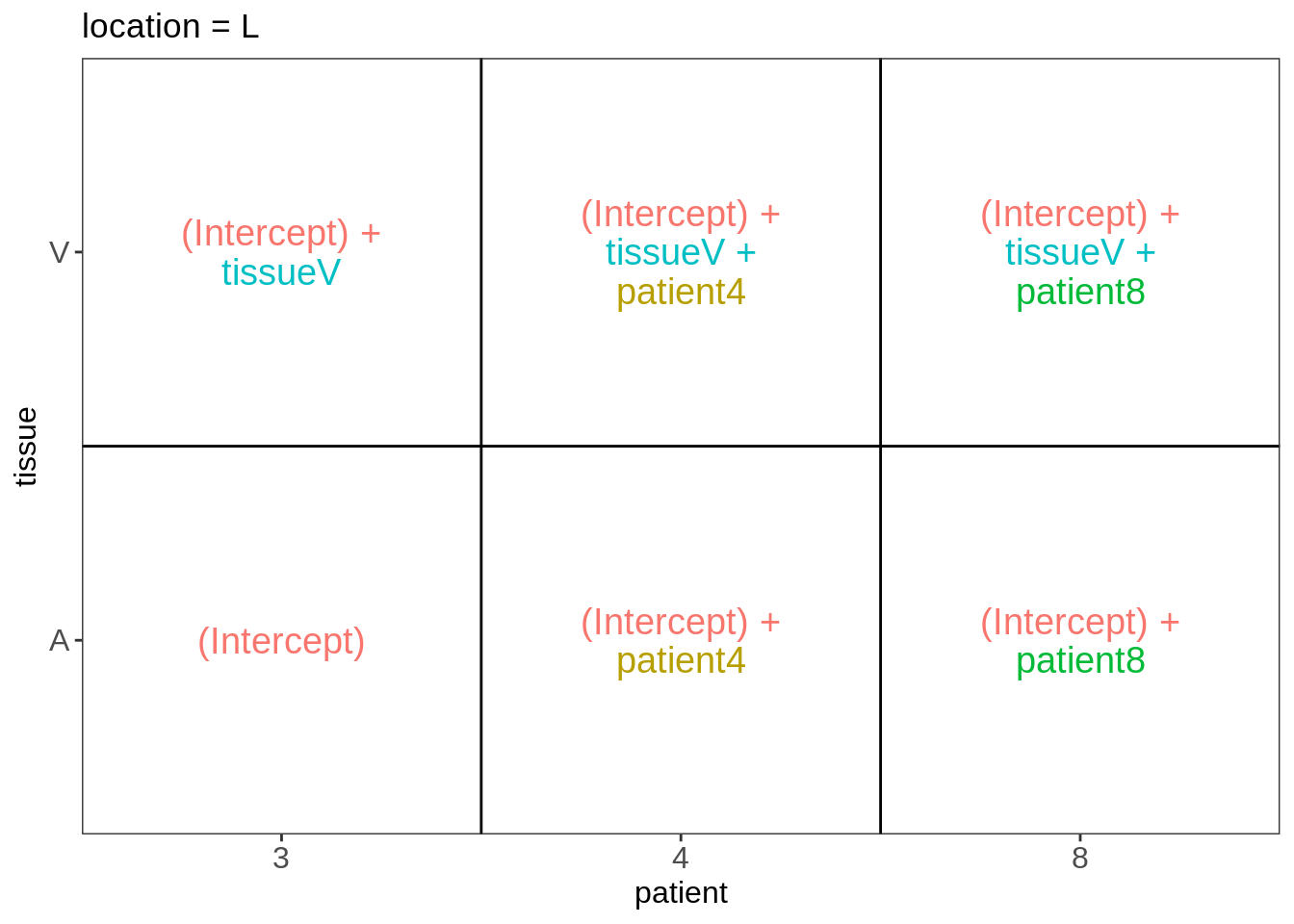
##
## $`location = R`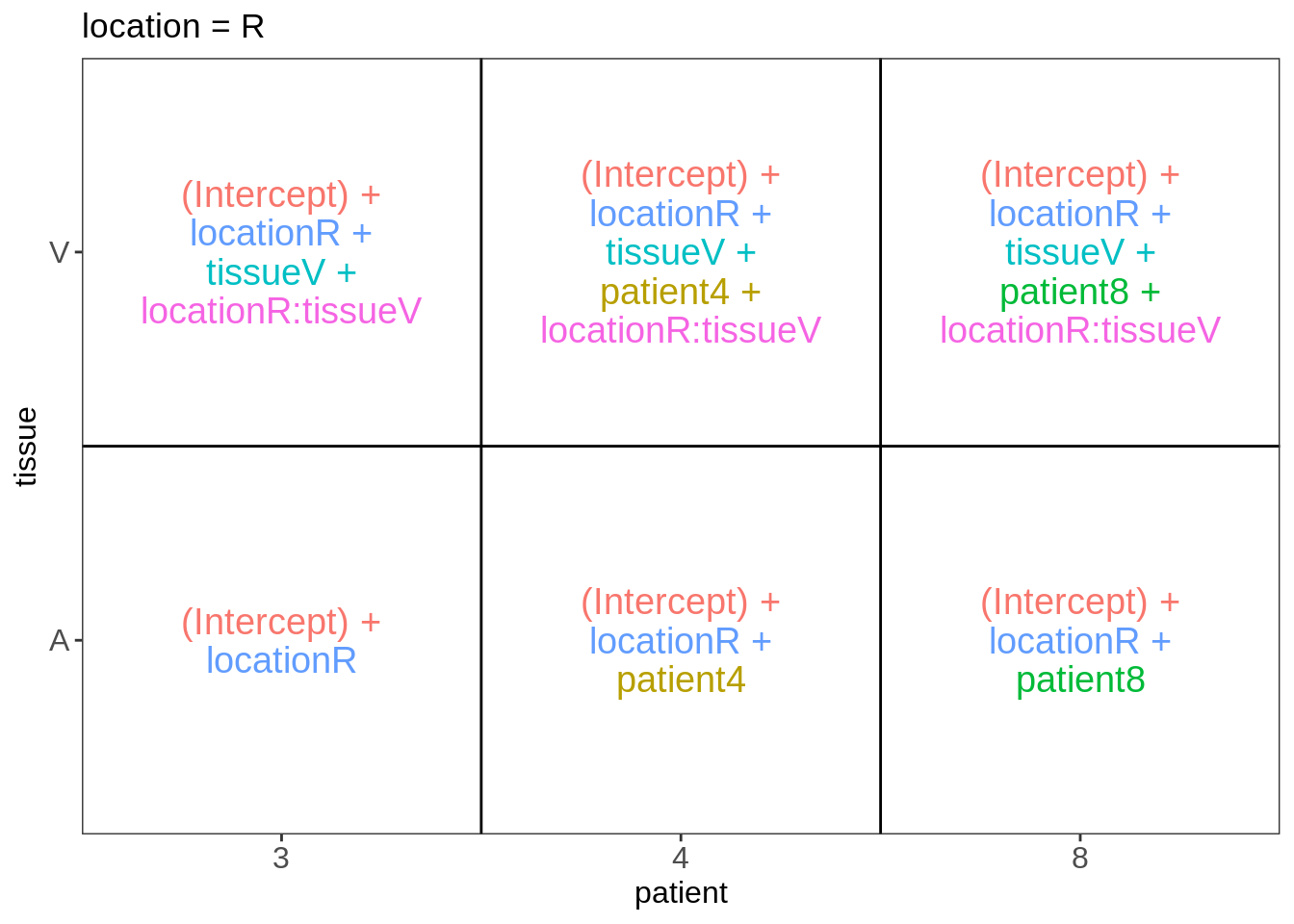
design <- model.matrix(~location*tissue + patient, data = colData(pe))
L <- makeContrast(
c(
"tissueV = 0",
"tissueV + locationR:tissueV = 0",
"tissueV + 0.5*locationR:tissueV = 0","locationR:tissueV = 0"),
parameterNames = colnames(design)
)
pe <- hypothesisTest(object = pe, i = "proteinRobust", contrast = L, overwrite=TRUE)4.3 Evaluate results contrast \(\log_2 FC_{V-A}^L\)
4.3.1 Volcano-plot
volcanoLeft <- ggplot(rowData(pe[["proteinRobust"]])$"tissueV",
aes(x = logFC, y = -log10(pval), color = adjPval < 0.05)) +
geom_point(cex = 2.5) +
scale_color_manual(values = alpha(c("black", "red"), 0.5)) + theme_minimal()
volcanoLeft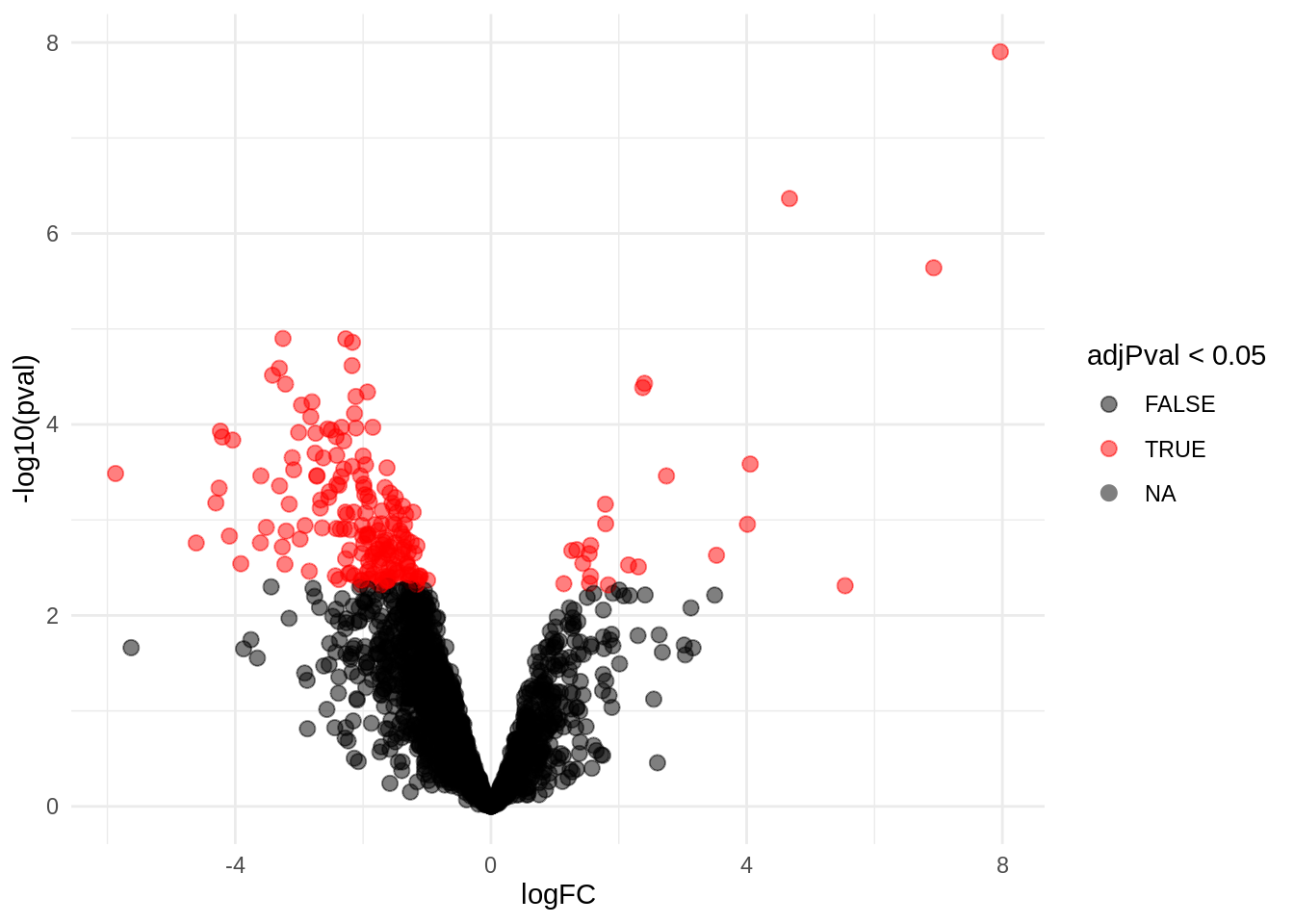
4.3.2 Heatmap
We first select the names of the proteins that were declared signficant.
sigNamesLeft <- rowData(pe[["proteinRobust"]])$tissueV %>%
rownames_to_column("proteinRobust") %>%
filter(adjPval<0.05) %>%
pull(proteinRobust)
heatmap(assay(pe[["proteinRobust"]])[sigNamesLeft, ])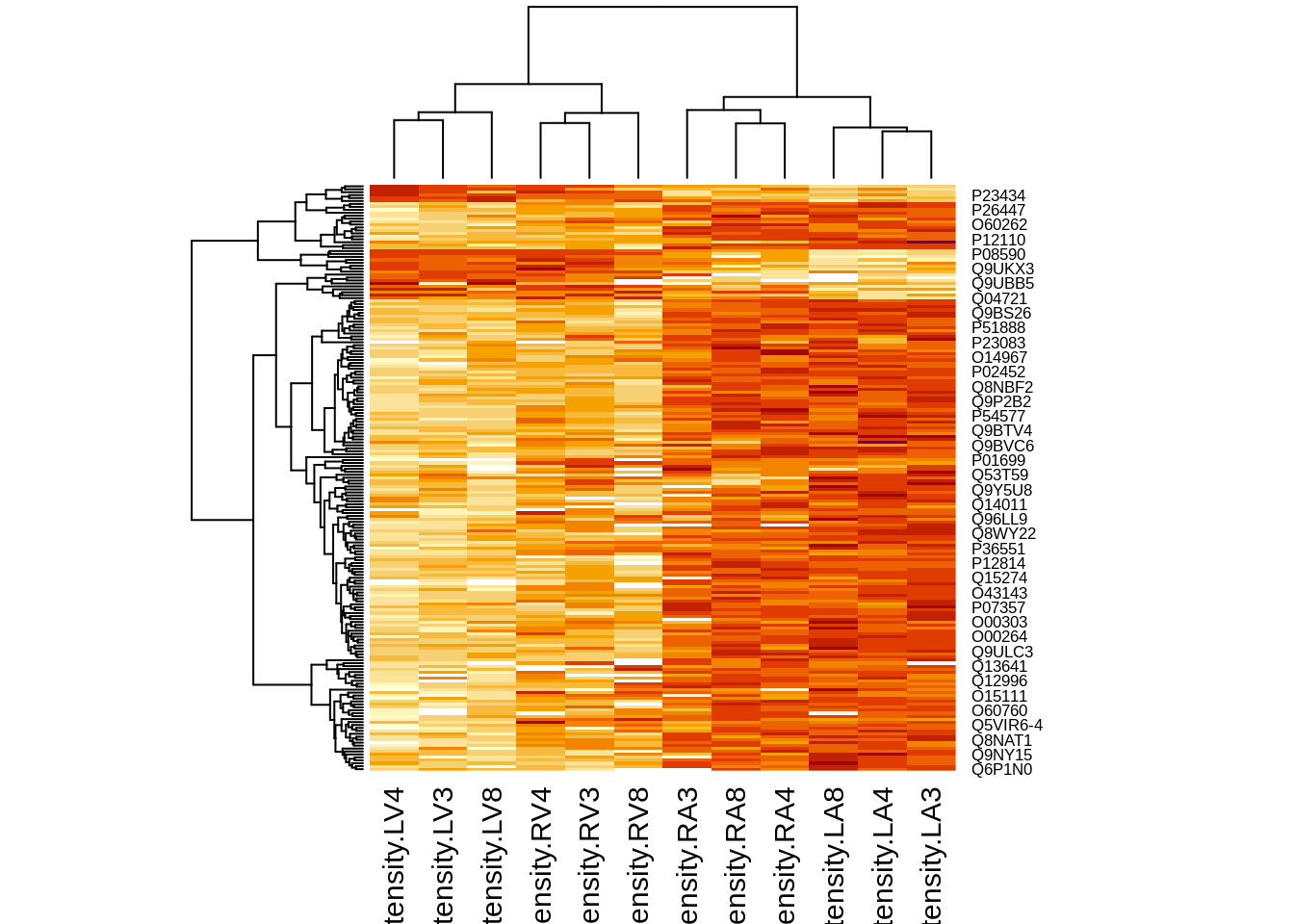
There are 199 proteins significantly differentially expressed at the 5% FDR level.
rowData(pe[["proteinRobust"]])$tissueV %>%
cbind(.,rowData(pe[["proteinRobust"]])$Protein.names) %>%
na.exclude %>%
filter(adjPval<0.05) %>%
arrange(pval) %>%
head(10) %>%
knitr::kable(.) | logFC | se | df | t | pval | adjPval | rowData(pe[[“proteinRobust”]])$Protein.names | |
|---|---|---|---|---|---|---|---|
| P08590 | 7.967951 | 0.4374873 | 9.367147 | 18.212987 | 0.00e+00 | 0.0000254 | Myosin light chain 3 |
| P12883 | 4.669883 | 0.3716628 | 9.181668 | 12.564838 | 4.00e-07 | 0.0004379 | Myosin-7 |
| P10916 | 6.926109 | 0.5291456 | 7.367147 | 13.089231 | 2.30e-06 | 0.0015506 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform |
| Q6UWY5 | -3.253643 | 0.3828898 | 9.113260 | -8.497596 | 1.26e-05 | 0.0046705 | Olfactomedin-like protein 1 |
| O75368 | -2.271594 | 0.2715292 | 9.289260 | -8.365932 | 1.27e-05 | 0.0046705 | SH3 domain-binding glutamic acid-rich-like protein |
| P46821 | -2.166880 | 0.2632626 | 9.367147 | -8.230871 | 1.38e-05 | 0.0046705 | Microtubule-associated protein 1B;MAP1B heavy chain;MAP1 light chain LC1 |
| O95865 | -2.173219 | 0.2826203 | 9.367147 | -7.689536 | 2.42e-05 | 0.0065478 | N(G),N(G)-dimethylarginine dimethylaminohydrolase 2 |
| Q8N474 | -3.309023 | 0.3804443 | 7.897481 | -8.697787 | 2.58e-05 | 0.0065478 | Secreted frizzled-related protein 1 |
| Q9ULL5-3 | -3.416540 | 0.3998973 | 7.846127 | -8.543546 | 3.05e-05 | 0.0068882 | Proline-rich protein 12 |
| P14854 | 2.401913 | 0.3057866 | 8.431469 | 7.854864 | 3.71e-05 | 0.0069621 | Cytochrome c oxidase subunit 6B1 |
4.4 Evaluate results contrast \(\log_2 FC_{V-A}^R\)
4.4.1 Volcano-plot
volcanoRight <- ggplot(rowData(pe[["proteinRobust"]])$"tissueV + locationR:tissueV",
aes(x = logFC, y = -log10(pval), color = adjPval < 0.05)) +
geom_point(cex = 2.5) +
scale_color_manual(values = alpha(c("black", "red"), 0.5)) + theme_minimal()
volcanoRight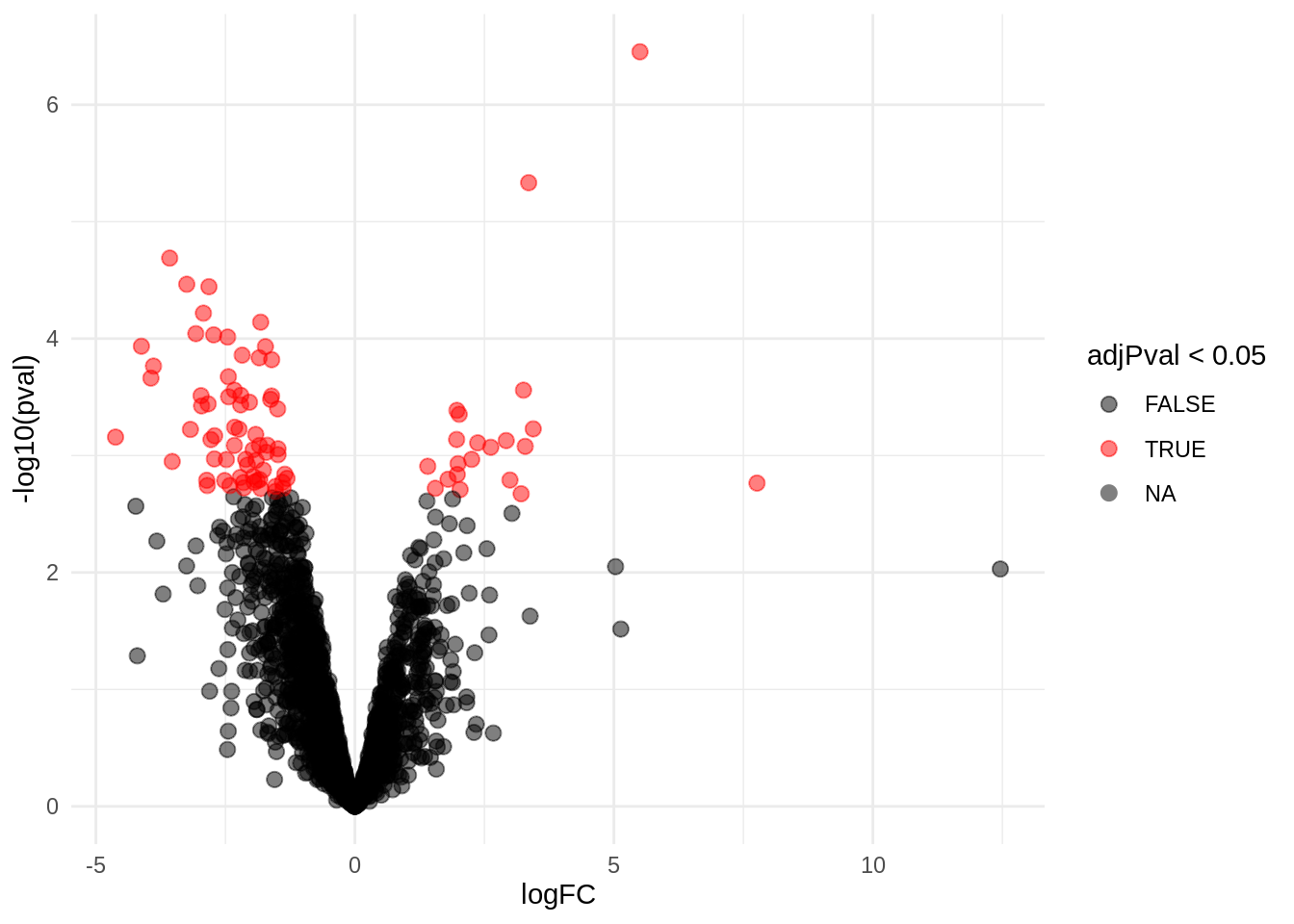
4.4.2 Heatmap
We first select the names of the proteins that were declared signficant.
sigNamesRight <- rowData(pe[["proteinRobust"]])$"tissueV + locationR:tissueV" %>%
rownames_to_column("proteinRobust") %>%
filter(adjPval<0.05) %>%
pull(proteinRobust)
heatmap(assay(pe[["proteinRobust"]])[sigNamesRight, ])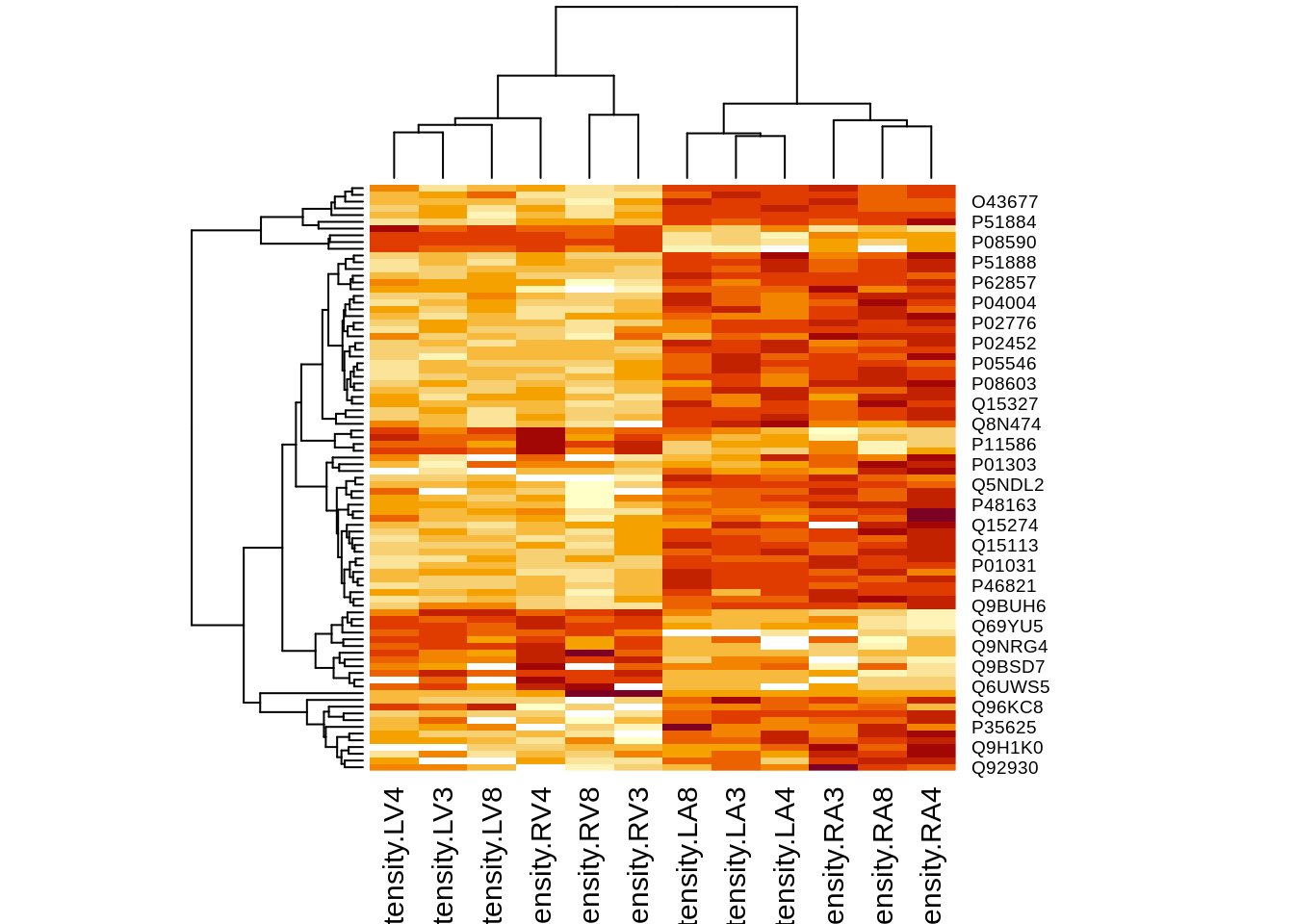
There are 87 proteins significantly differentially expressed at the 5% FDR level.
rowData(pe[["proteinRobust"]])$"tissueV + locationR:tissueV" %>%
cbind(.,rowData(pe[["proteinRobust"]])$Protein.names) %>%
na.exclude %>%
filter(adjPval<0.05) %>%
arrange(pval) %>%
head(10) %>%
knitr::kable(.)| logFC | se | df | t | pval | adjPval | rowData(pe[[“proteinRobust”]])$Protein.names | |
|---|---|---|---|---|---|---|---|
| P08590 | 5.503838 | 0.4374873 | 9.367147 | 12.580565 | 4.00e-07 | 0.0007167 | Myosin light chain 3 |
| P06858 | 3.354674 | 0.3544630 | 9.249267 | 9.464102 | 4.60e-06 | 0.0047205 | Lipoprotein lipase |
| Q9ULD0 | -3.575906 | 0.3737222 | 7.367147 | -9.568355 | 2.05e-05 | 0.0138839 | 2-oxoglutarate dehydrogenase-like, mitochondrial |
| P35442 | -3.246460 | 0.4065828 | 8.367147 | -7.984746 | 3.43e-05 | 0.0146265 | Thrombospondin-2 |
| P02776 | -2.818363 | 0.3421511 | 7.975973 | -8.237189 | 3.60e-05 | 0.0146265 | Platelet factor 4;Platelet factor 4, short form |
| P21810 | -2.924783 | 0.4081159 | 8.756796 | -7.166550 | 6.06e-05 | 0.0197128 | Biglycan |
| O75368 | -1.819326 | 0.2697137 | 9.289260 | -6.745396 | 7.23e-05 | 0.0197128 | SH3 domain-binding glutamic acid-rich-like protein |
| A6NMZ7 | -3.070848 | 0.4709254 | 9.367147 | -6.520879 | 9.07e-05 | 0.0197128 | Collagen alpha-6(VI) chain |
| P54652 | -2.726681 | 0.4117048 | 9.078544 | -6.622904 | 9.29e-05 | 0.0197128 | Heat shock-related 70 kDa protein 2 |
| Q6UWY5 | -2.456874 | 0.3739352 | 9.113260 | -6.570320 | 9.70e-05 | 0.0197128 | Olfactomedin-like protein 1 |
4.5 Evaluate results average contrast \(\log_2 FC_{V-A}\)
4.5.1 Volcano-plot
volcanoAvg <- ggplot(rowData(pe[["proteinRobust"]])$"tissueV + 0.5 * locationR:tissueV",
aes(x = logFC, y = -log10(pval), color = adjPval < 0.05)) +
geom_point(cex = 2.5) +
scale_color_manual(values = alpha(c("black", "red"), 0.5)) + theme_minimal()
volcanoAvg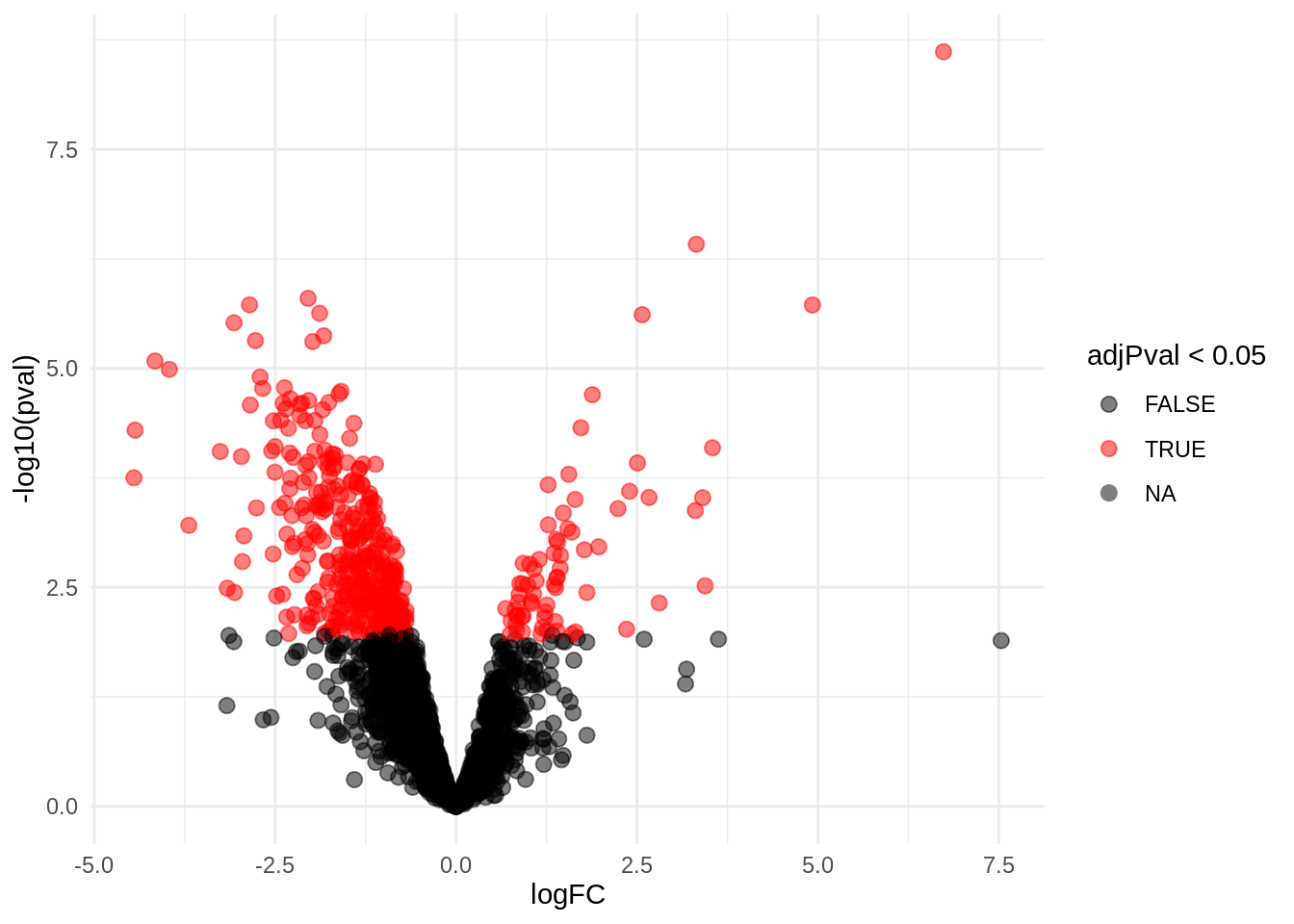
4.5.2 Heatmap
We first select the names of the proteins that were declared signficant.
sigNamesAvg <- rowData(pe[["proteinRobust"]])$"tissueV + 0.5 * locationR:tissueV" %>%
rownames_to_column("proteinRobust") %>%
filter(adjPval<0.05) %>%
pull(proteinRobust)
heatmap(assay(pe[["proteinRobust"]])[sigNamesAvg, ])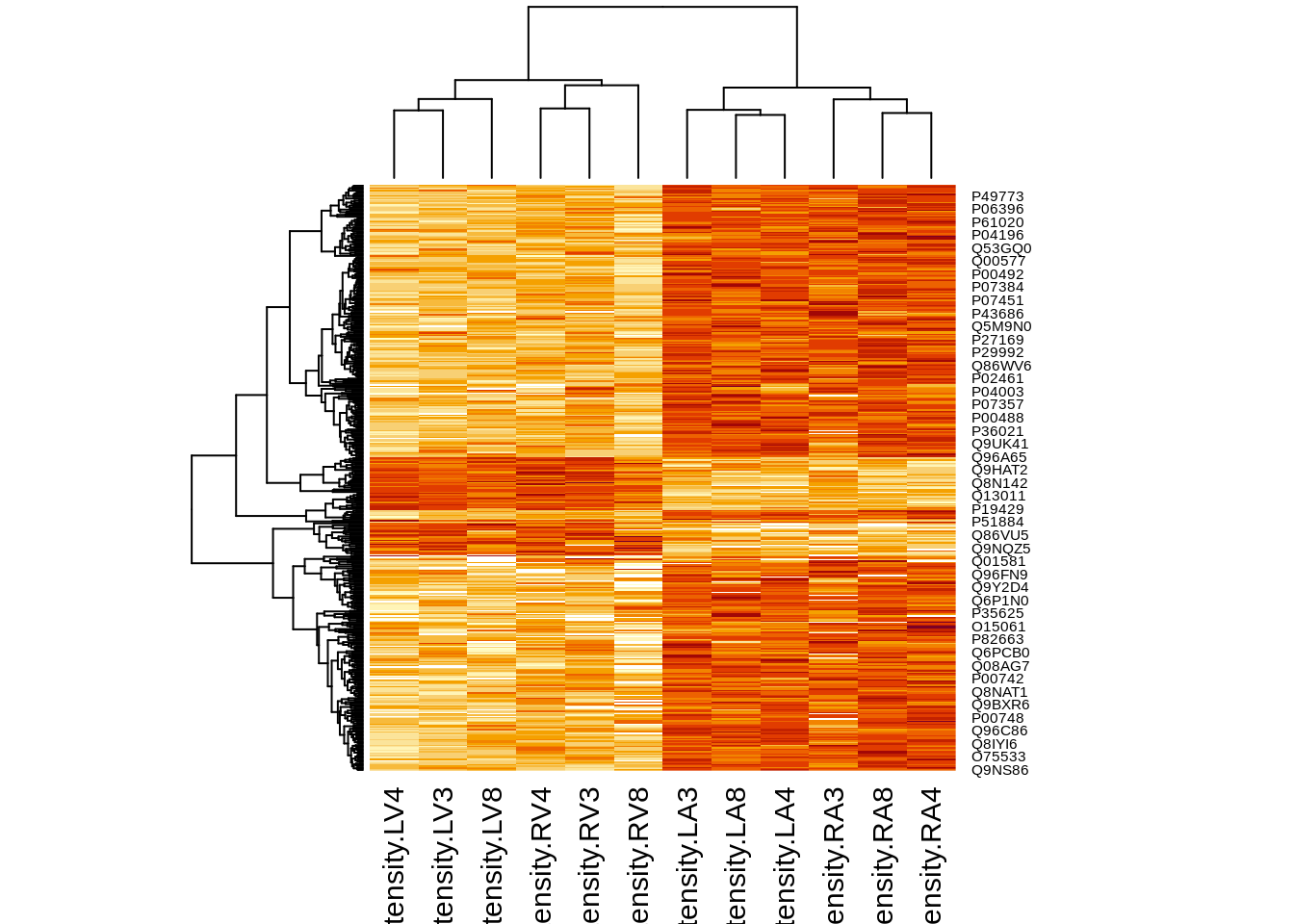
There are 449 proteins significantly differentially expressed at the 5% FDR level.
rowData(pe[["proteinRobust"]])$"tissueV + 0.5 * locationR:tissueV" %>%
cbind(.,rowData(pe[["proteinRobust"]])$Protein.names) %>%
na.exclude %>%
filter(adjPval<0.05) %>%
arrange(pval) %>%
head(10) %>%
knitr::kable(.) | logFC | se | df | t | pval | adjPval | rowData(pe[[“proteinRobust”]])$Protein.names | |
|---|---|---|---|---|---|---|---|
| P08590 | 6.735894 | 0.3093503 | 9.367147 | 21.77433 | 0.0e+00 | 0.0000049 | Myosin light chain 3 |
| P12883 | 3.319491 | 0.2606314 | 9.181668 | 12.73634 | 4.0e-07 | 0.0003891 | Myosin-7 |
| O75368 | -2.045460 | 0.1913593 | 9.289260 | -10.68911 | 1.6e-06 | 0.0007082 | SH3 domain-binding glutamic acid-rich-like protein |
| Q6UWY5 | -2.855258 | 0.2675968 | 9.113260 | -10.67000 | 1.9e-06 | 0.0007082 | Olfactomedin-like protein 1 |
| P10916 | 4.923642 | 0.3661150 | 7.367147 | 13.44835 | 1.9e-06 | 0.0007082 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform |
| P46821 | -1.886709 | 0.1861547 | 9.367147 | -10.13517 | 2.4e-06 | 0.0007082 | Microtubule-associated protein 1B;MAP1B heavy chain;MAP1 light chain LC1 |
| P06858 | 2.571933 | 0.2519480 | 9.249267 | 10.20819 | 2.4e-06 | 0.0007082 | Lipoprotein lipase |
| P21810 | -3.069662 | 0.2931217 | 8.756796 | -10.47231 | 3.0e-06 | 0.0007662 | Biglycan |
| P05546 | -1.829841 | 0.1916993 | 9.271661 | -9.54537 | 4.2e-06 | 0.0009134 | Heparin cofactor 2 |
| P35442 | -2.773755 | 0.2689292 | 8.367147 | -10.31407 | 4.8e-06 | 0.0009134 | Thrombospondin-2 |
4.6 Interaction
4.6.1 Volcano-plot
volcanoInt <- ggplot(rowData(pe[["proteinRobust"]])$"locationR:tissueV",
aes(x = logFC, y = -log10(pval), color = adjPval < 0.05)) +
geom_point(cex = 2.5) +
scale_color_manual(values = alpha(c("black", "red"), 0.5)) + theme_minimal()
volcanoInt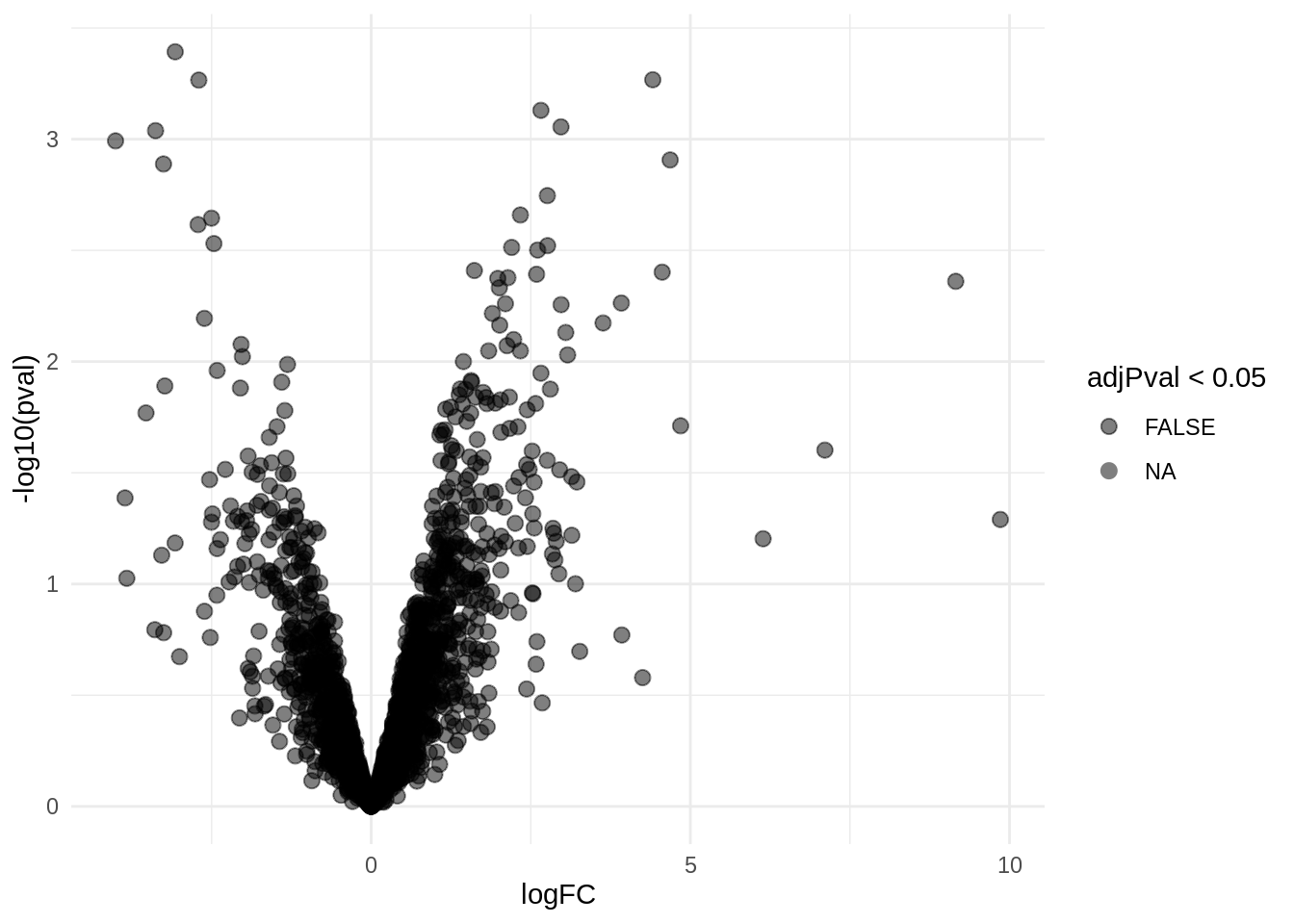
4.6.2 Heatmap
There were no genes significant at the 5% FDR level. We return the top 25 genes.
sigNamesInt <- rowData(pe[["proteinRobust"]])$"locationR:tissueV" %>%
rownames_to_column("proteinRobust") %>%
filter(adjPval<0.05) %>%
pull(proteinRobust)
hlp <- order((rowData(pe[["proteinRobust"]])$"locationR:tissueV")[,"adjPval"])[1:25]
heatmap(assay(pe[["proteinRobust"]])[hlp, ])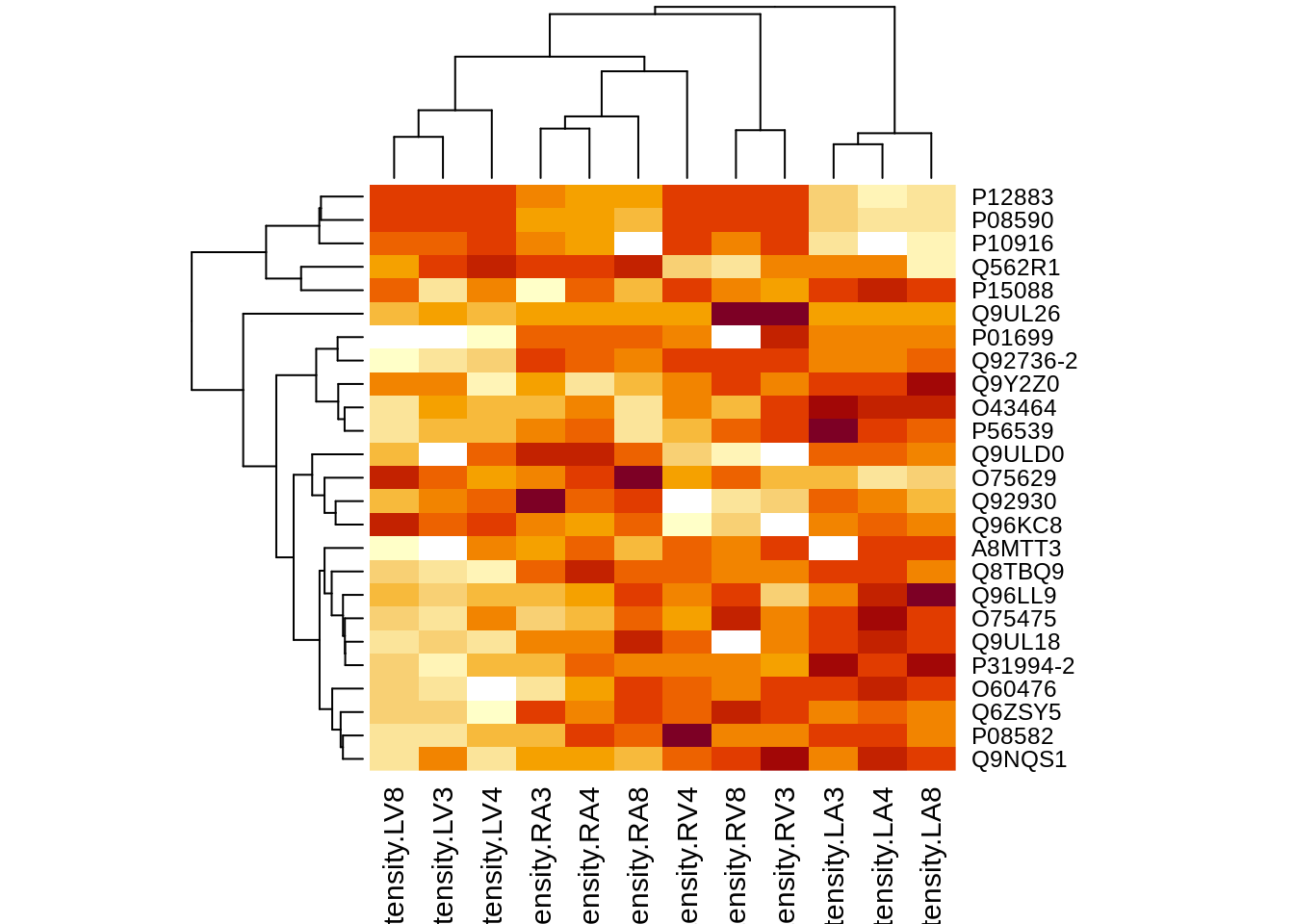
There are 0 proteins significantly differentially expressed at the 5% FDR level.
rowData(pe[["proteinRobust"]])$"locationR:tissueV" %>%
cbind(.,rowData(pe[["proteinRobust"]])$Protein.names) %>%
na.exclude %>%
filter(adjPval<0.05) %>%
head(10) %>%
knitr::kable(.) | logFC | se | df | t | pval | adjPval | rowData(pe[[“proteinRobust”]])$Protein.names |
|---|