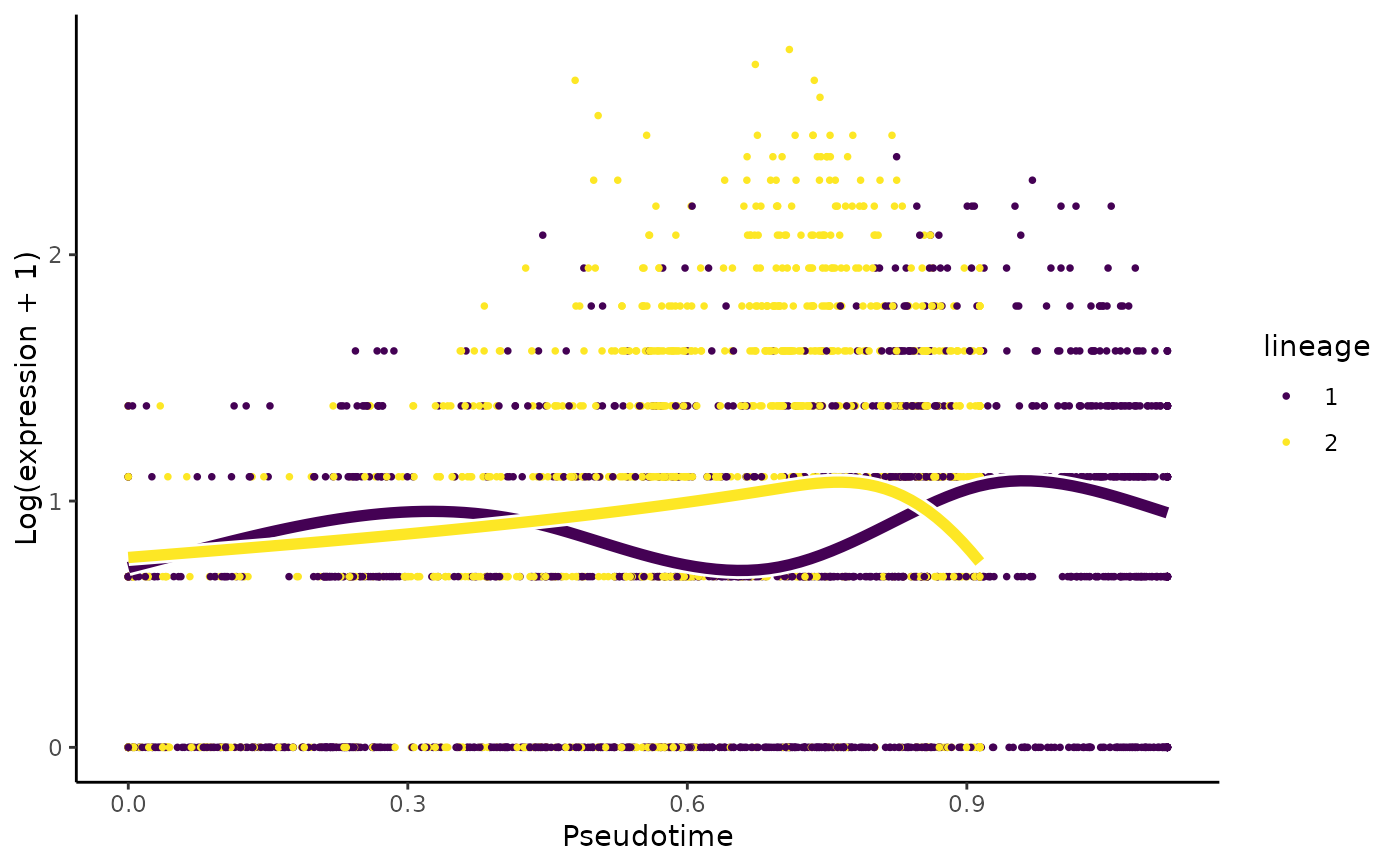
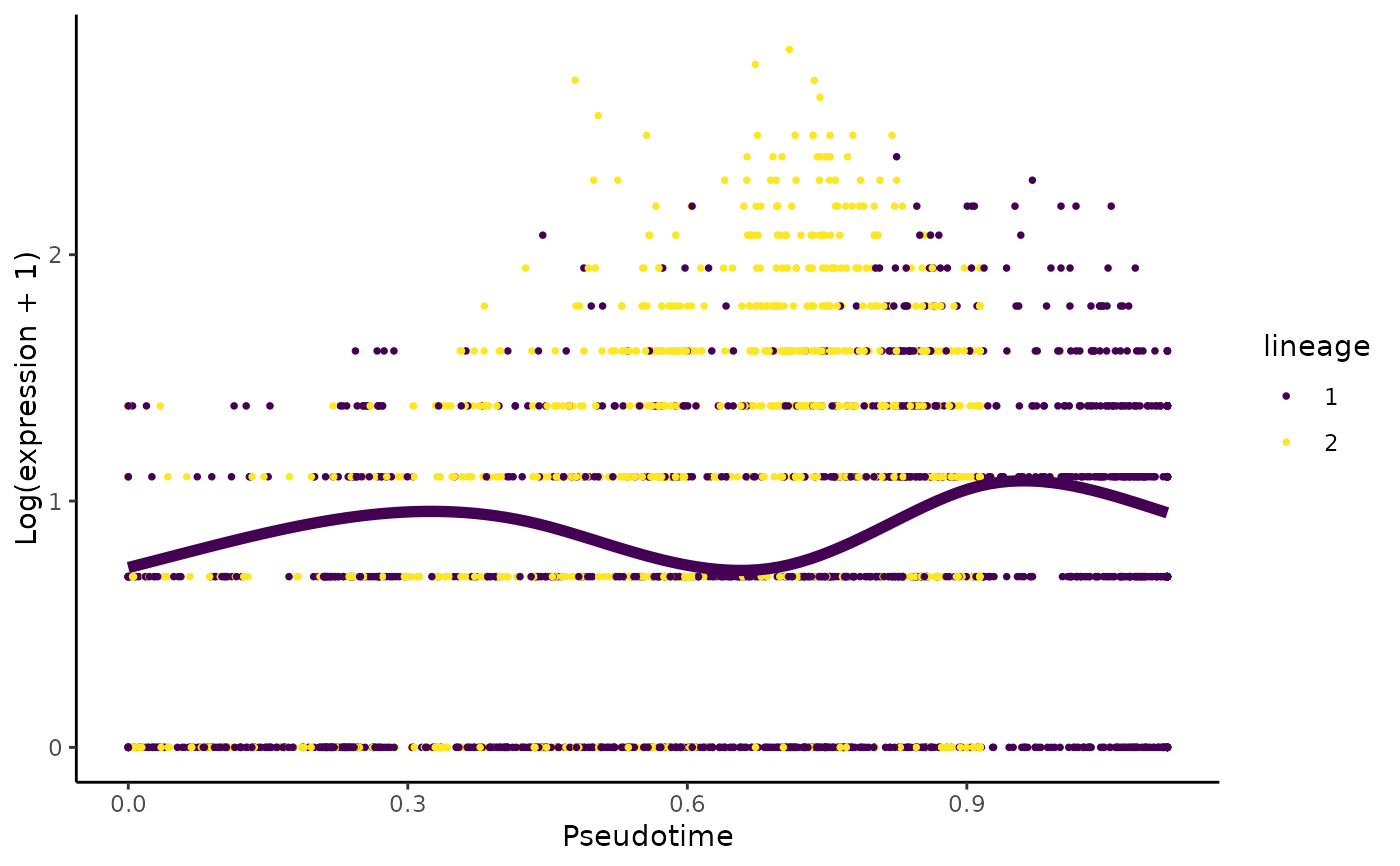
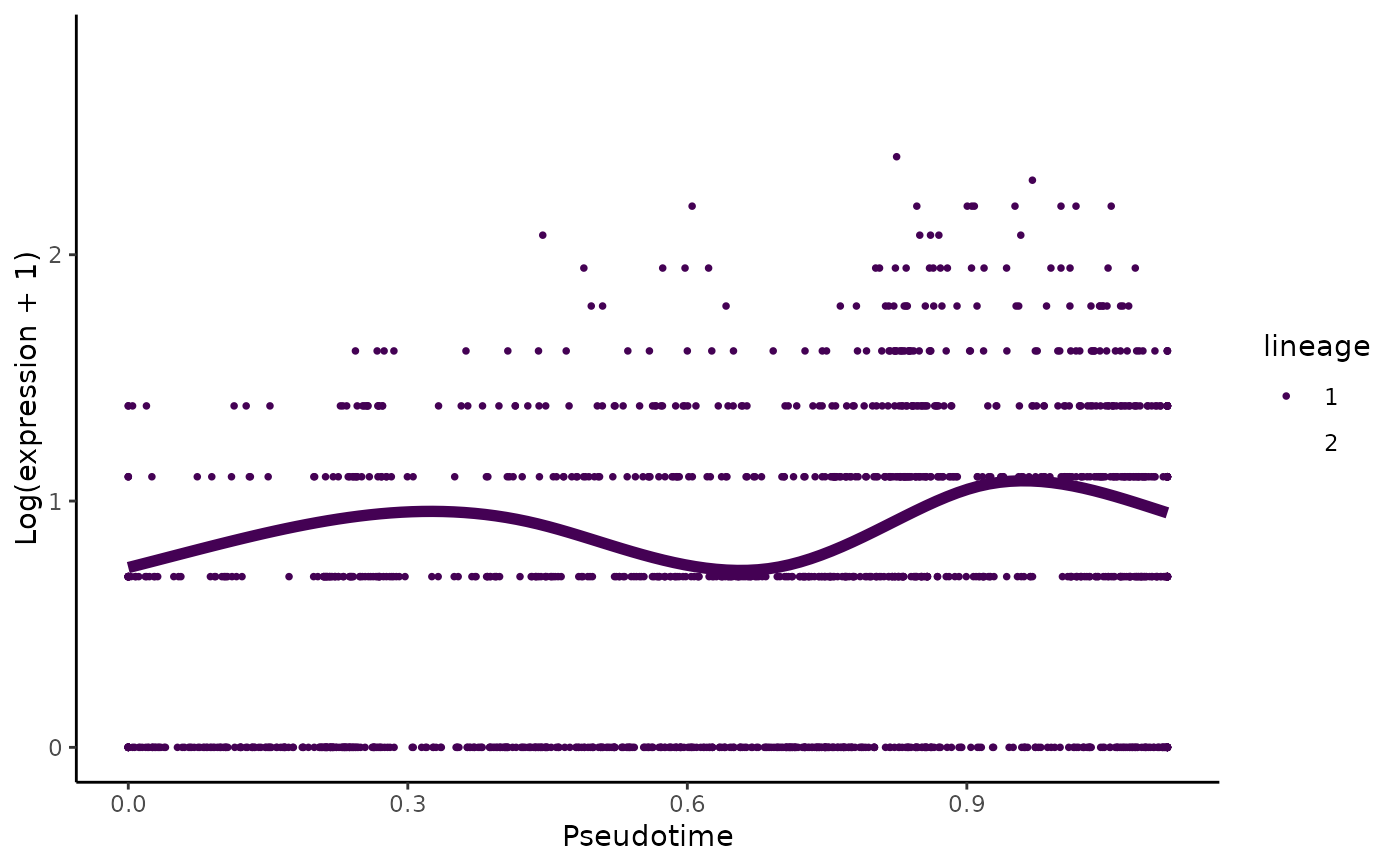
Plot the log-transformed counts and the fitted values for a particular gene along all lineages
Source:R/AllGenerics.R, R/plotSmoothers.R
plotSmoothers.RdPlot the smoothers estimated by tradeSeq.
plotSmoothers(models, ...) # S4 method for gam plotSmoothers( models, nPoints = 100, lwd = 2, size = 2/3, xlab = "Pseudotime", ylab = "Log(expression + 1)", border = TRUE, alpha = 1, sample = 1 ) # S4 method for SingleCellExperiment plotSmoothers( models, counts, gene, nPoints = 100, lwd = 2, size = 2/3, xlab = "Pseudotime", ylab = "Log(expression + 1)", border = TRUE, alpha = 1, sample = 1, pointCol = NULL, curvesCols = NULL, plotLineages = TRUE )
Arguments
| models | Either the |
|---|---|
| ... | parameters including: |
| nPoints | The number of points used to extrapolate the fit. Defaults to 100. |
| lwd | Line width of the smoother. Passed to |
| size | Character expansion of the data points. Passed to |
| xlab | x-axis label. Passed to |
| ylab | y-axis label. Passed to |
| border | Logical: should a white border be drawn around the mean smoother. |
| alpha | Numeric between 0 and 1, determines the transparency of data points,
see |
| sample | Numeric between 0 and 1, use to subsample the cells when there are too many so that it can plot faster. |
| counts | The matrix of gene expression counts. |
| gene | Gene name or row in count matrix of gene to plot. |
| pointCol | Plotting colors for each cell. Can be either character vector of
length 1, denoting a variable in the |
| curvesCols | Plotting colors for each curve Should be a list of colors of the exact same length as the number of curves, i.e. the number of lineages (if there is no conditions) or the number of lineages by the number of conditions. In the second case, the colors are grouped by condition (lineage 1 - condition 1, lineage 1 - condition 2,...). |
| plotLineages | Logical, should the mean smoothers for each lineage be plotted? |
Value
A ggplot object
Examples
set.seed(8) data(crv, package="tradeSeq") data(countMatrix, package="tradeSeq") counts <- as.matrix(countMatrix) sce <- fitGAM(counts = counts, sds = crv, nknots = 5) plotSmoothers(sce, counts, rownames(counts)[1])# Show only first lineage curve curvesCols <- c("#440154FF", "transparent") plotSmoothers(sce, counts, rownames(counts)[1], curvesCols = curvesCols, border = FALSE)# Show only first curve and cells assigned to first lineage plotSmoothers(sce, counts, rownames(counts)[1], curvesCols = curvesCols, border = FALSE) + ggplot2::scale_color_manual(values = curvesCols)#> #>