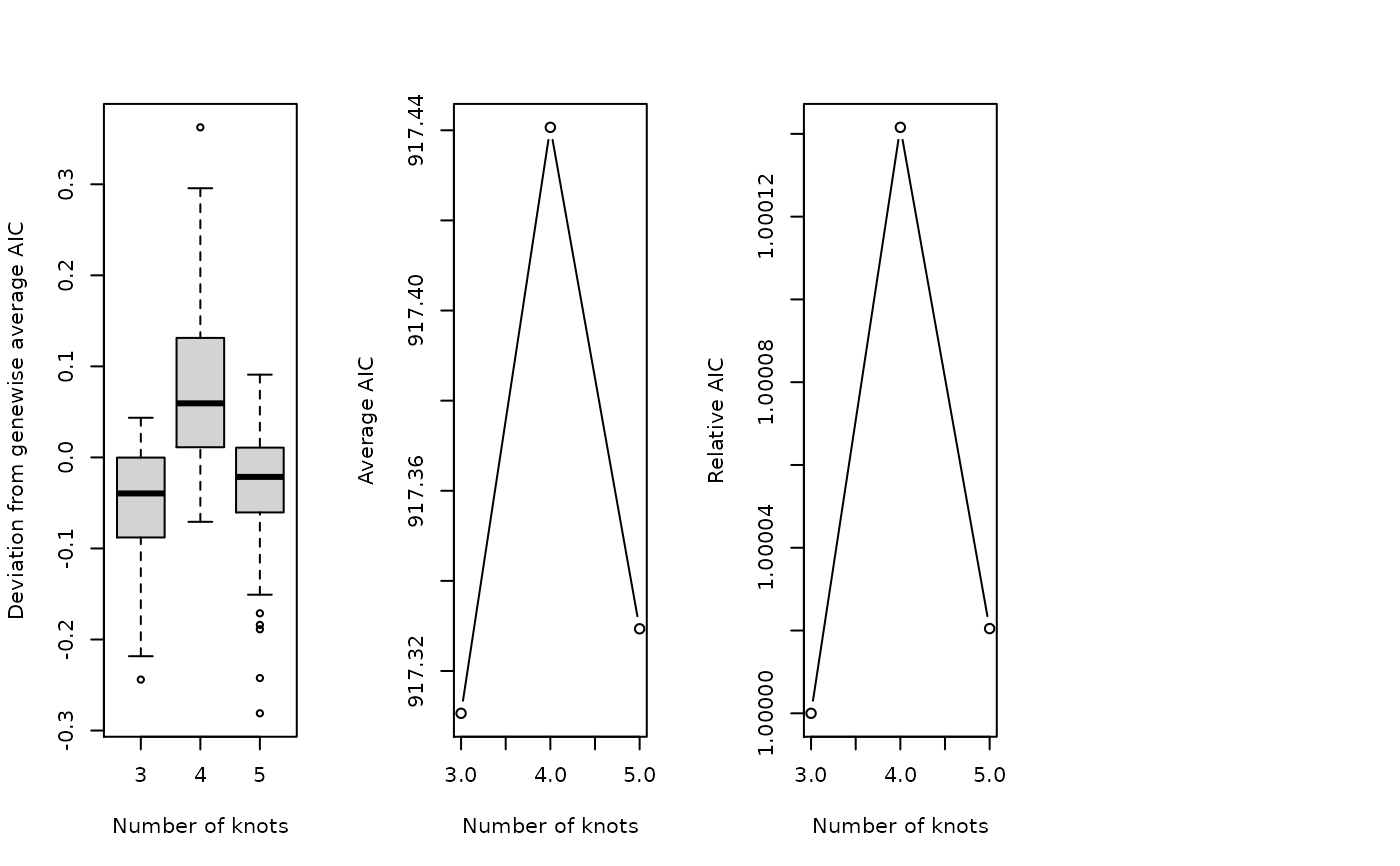Evaluate the optimal number of knots required for fitGAM.
Source:R/AllGenerics.R, R/evaluateK.R
evaluateK.RdEvaluate the optimal number of knots required for fitGAM.
Evaluate an appropriate number of knots.
evaluateK(counts, ...) # S4 method for matrix evaluateK( counts, k = 3:10, nGenes = 500, sds = NULL, pseudotime = NULL, cellWeights = NULL, U = NULL, conditions = NULL, plot = TRUE, weights = NULL, offset = NULL, aicDiff = 2, verbose = TRUE, parallel = FALSE, BPPARAM = BiocParallel::bpparam(), control = mgcv::gam.control(), family = "nb", gcv = FALSE, ... ) # S4 method for dgCMatrix evaluateK( counts, k = 3:10, nGenes = 500, sds = NULL, pseudotime = NULL, cellWeights = NULL, plot = TRUE, U = NULL, weights = NULL, offset = NULL, aicDiff = 2, verbose = TRUE, conditions = NULL, control = mgcv::gam.control(), parallel = FALSE, BPPARAM = BiocParallel::bpparam(), family = "nb", gcv = FALSE, ... ) # S4 method for SingleCellExperiment evaluateK( counts, k = 3:10, nGenes = 500, sds = NULL, pseudotime = NULL, cellWeights = NULL, plot = TRUE, U = NULL, weights = NULL, offset = NULL, aicDiff = 2, verbose = TRUE, conditions = NULL, parallel = FALSE, BPPARAM = BiocParallel::bpparam(), control = mgcv::gam.control(), family = "nb", gcv = FALSE, ... ) # S4 method for CellDataSet evaluateK( counts, k = 3:10, nGenes = 500, sds = NULL, pseudotime = NULL, cellWeights = NULL, plot = TRUE, U = NULL, weights = NULL, offset = NULL, aicDiff = 2, verbose = TRUE, conditions = NULL, parallel = FALSE, BPPARAM = BiocParallel::bpparam(), control = mgcv::gam.control(), family = "nb", gcv = FALSE, ... )
Arguments
| counts | The count matrix, genes in rows and cells in columns. |
|---|---|
| ... | parameters including: |
| k | The range of knots to evaluate. `3:10` by default. See details. |
| nGenes | The number of genes to use in the evaluation. Genes will be randomly selected. 500 by default. |
| sds | Slingshot object containing the lineages. |
| pseudotime | a matrix of pseudotime values, each row represents a cell and each column represents a lineage. |
| cellWeights | a matrix of cell weights defining the probability that a cell belongs to a particular lineage. Each row represents a cell and each column represents a lineage. |
| U | The design matrix of fixed effects. The design matrix should not contain an intercept to ensure identifiability. |
| conditions | This argument is in beta phase and should be used carefully. If each lineage consists of multiple conditions, this argument can be used to specify the conditions. tradeSeq will then fit a condition-specific smoother for every lineage. |
| plot | Whether to display diagnostic plots. Default to |
| weights | Optional: a matrix of weights with identical dimensions
as the |
| offset | Optional: the offset, on log-scale. If NULL, TMM is used to
account for differences in sequencing depth, see |
| aicDiff | Used for selecting genes with significantly varying AIC values over the range of evaluated knots to make the barplot output. Default is set to 2, meaning that only genes whose AIC range is larger than 2 will be used to check for the optimal number of knots through the barplot visualization that is part of the output of this function. |
| verbose | logical, should progress be verbose? |
| parallel | Logical, defaults to FALSE. Set to TRUE if you want to parallellize the fitting. |
| BPPARAM | object of class |
| control | Control object for GAM fitting, see |
| family | The distribution assumed, currently only |
| gcv | (In development). Logical, should a GCV score also be returned? |
Value
A plot of average AIC value over the range of selected knots, and a matrix of AIC and GCV values for the selected genes (rows) and the range of knots (columns).
Details
The number of parameter to evaluate (and therefore the runtime) scales in
k * the number of lineages. Morevoer, we have found that, in practice,
values of k above 12-15 rarely lead to improved result, not matter the
complexity of the trajectory being considered. As such, we recommand that user
proceed with care when setting k to value higher than 15.
Examples
## This is an artificial example, please check the vignette for a realistic one. set.seed(8) data(sds, package="tradeSeq") loadings <- matrix(runif(2000*2, -2, 2), nrow = 2, ncol = 2000) counts <- round(abs(t(slingshot::reducedDim(sds) %*% loadings))) + 100 aicK <- evaluateK(counts = counts, sds = sds, nGenes = 100, k = 3:5, verbose = FALSE)