Plot function to visualize differential transcript usage (DTU)
plotDTU(
object,
contrast,
groups,
coefficients,
summaryStat = "model",
transcripts = NULL,
genes = NULL,
top.n = 6
)Arguments
- object
A `SummarizedExperiment` containing the models and results of the DTU analysis as obtained by the `fitDTU` and `testDTU` function from this `satuRn` package, respectively.
- contrast
Specifies the specific contrast for which the visualization should be returned. This should be one of the column names of the contrast matrix that was provided to the `testDTU` function.
- groups
A `list` containing two character vectors. Each character vector contains the names (sample names or cell names) of the respective groups in the target contrast.
- coefficients
A `list` containing two numeric vectors. Each numeric vector specifies the model coefficient of the corresponding groups in the selected contrast.
- summaryStat
Which summary statistic for the relative usage of the transcript should be displayed. `Character` or `character vector`, must be any of following summary statistics; model (default), mean or median.
- transcripts
A `character` or `character vector` of transcript IDs, to specify which transcripts should be visualized. Can be used together with `genes`. If not specified, `plotDTU` will check if the `genes` slot is specified.
- genes
A single `character` or `character vector` of gene IDs, to specify the genes for which the individual transcripts should be visualized. Can be used together with `transcripts`. If not specified, `plotDTU` will check if the `transcripts` slot is specified.
- top.n
A `numeric` value. If neither `transcripts` nor `genes` was was specified, this argument leads to the visualization of the `n` most significant DTU transcripts in the contrast. Defaults to 6 transcripts.
Value
A ggplot object that can be directly displayed in the current R session or stored in a list.
Examples
data(sumExp_example, package = "satuRn")
library(SummarizedExperiment)
#> Loading required package: MatrixGenerics
#> Loading required package: matrixStats
#>
#> Attaching package: ‘MatrixGenerics’
#> The following objects are masked from ‘package:matrixStats’:
#>
#> colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
#> colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
#> colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
#> colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
#> colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
#> colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
#> colWeightedMeans, colWeightedMedians, colWeightedSds,
#> colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
#> rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
#> rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
#> rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
#> rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
#> rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
#> rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
#> rowWeightedSds, rowWeightedVars
#> Loading required package: GenomicRanges
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#>
#> Attaching package: ‘BiocGenerics’
#> The following objects are masked from ‘package:stats’:
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from ‘package:base’:
#>
#> Filter, Find, Map, Position, Reduce, anyDuplicated, aperm, append,
#> as.data.frame, basename, cbind, colnames, dirname, do.call,
#> duplicated, eval, evalq, get, grep, grepl, intersect, is.unsorted,
#> lapply, mapply, match, mget, order, paste, pmax, pmax.int, pmin,
#> pmin.int, rank, rbind, rownames, sapply, setdiff, sort, table,
#> tapply, union, unique, unsplit, which.max, which.min
#> Loading required package: S4Vectors
#>
#> Attaching package: ‘S4Vectors’
#> The following objects are masked from ‘package:base’:
#>
#> I, expand.grid, unname
#> Loading required package: IRanges
#> Loading required package: GenomeInfoDb
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: ‘Biobase’
#> The following object is masked from ‘package:MatrixGenerics’:
#>
#> rowMedians
#> The following objects are masked from ‘package:matrixStats’:
#>
#> anyMissing, rowMedians
sumExp <- fitDTU(
object = sumExp_example,
formula = ~ 0 + group,
parallel = FALSE,
BPPARAM = BiocParallel::bpparam(),
verbose = TRUE
)
group <- as.factor(colData(sumExp)$group)
design <- model.matrix(~ 0 + group)
colnames(design) <- levels(group)
L <- matrix(0, ncol = 2, nrow = ncol(design))
rownames(L) <- colnames(design)
colnames(L) <- c("Contrast1", "Contrast2")
L[c("VISp.L5_IT_VISp_Hsd11b1_Endou", "ALM.L5_IT_ALM_Tnc"), 1] <- c(1, -1)
L[c("VISp.L5_IT_VISp_Hsd11b1_Endou",
"ALM.L5_IT_ALM_Tmem163_Dmrtb1"), 2] <- c(1, -1)
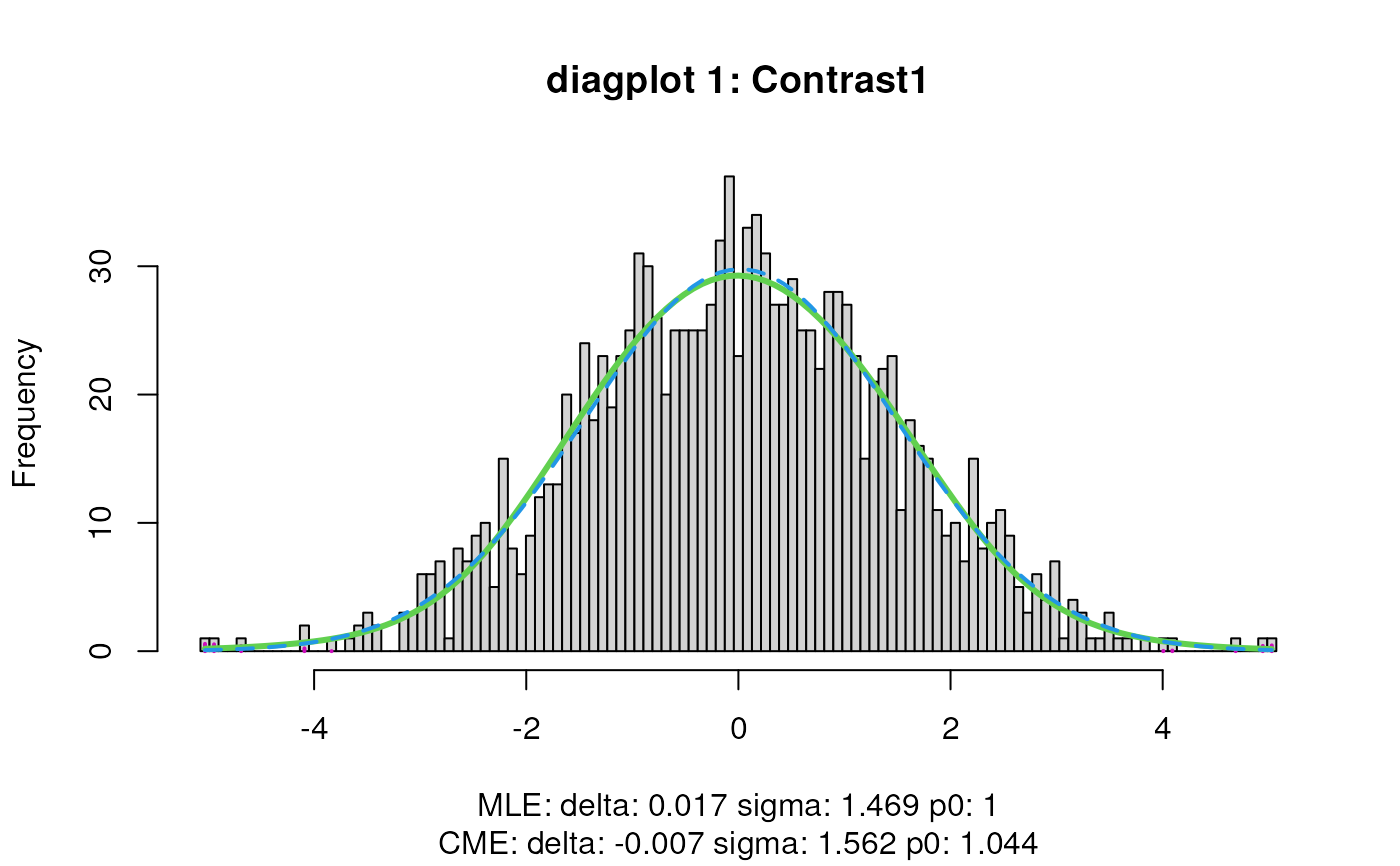
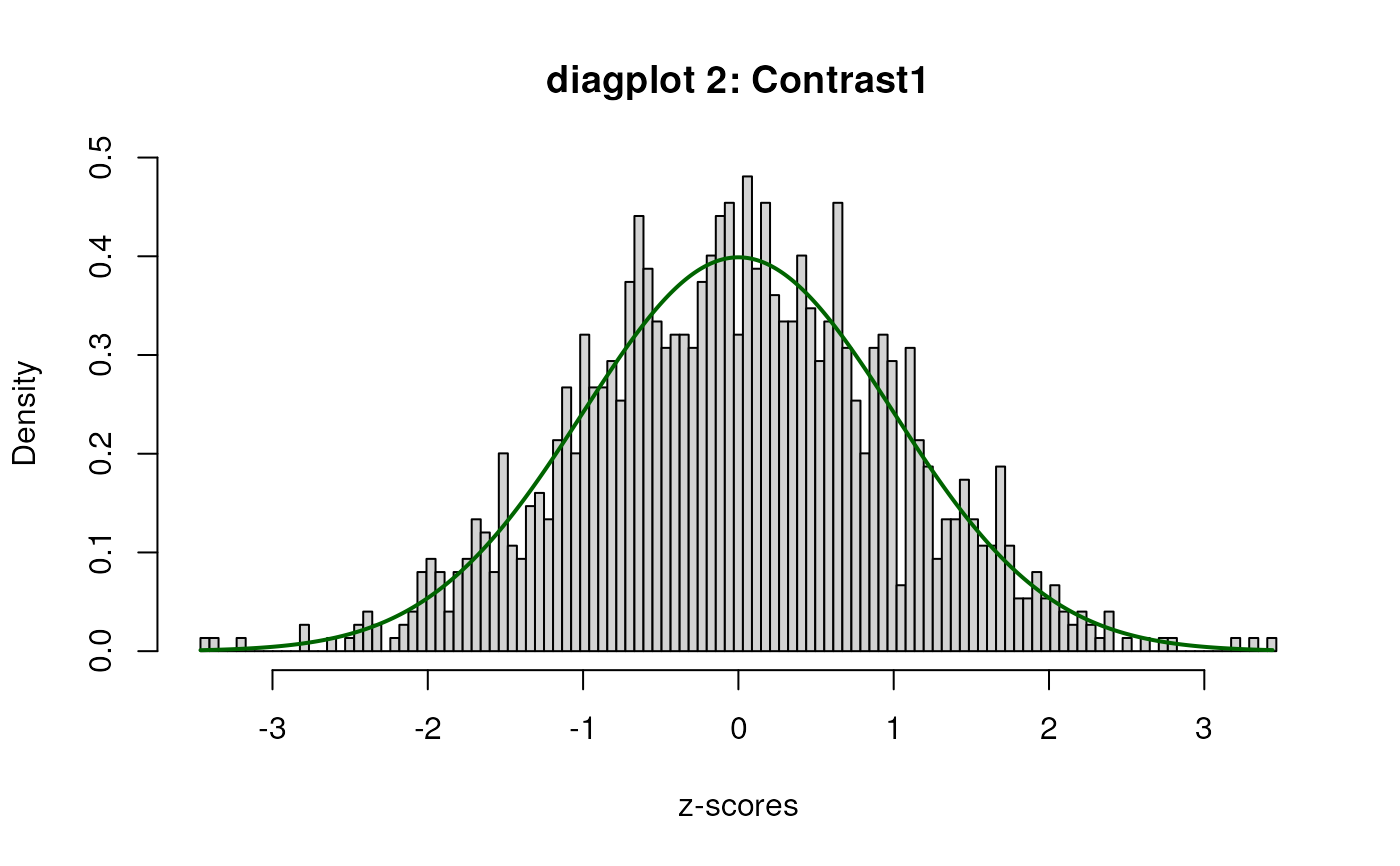
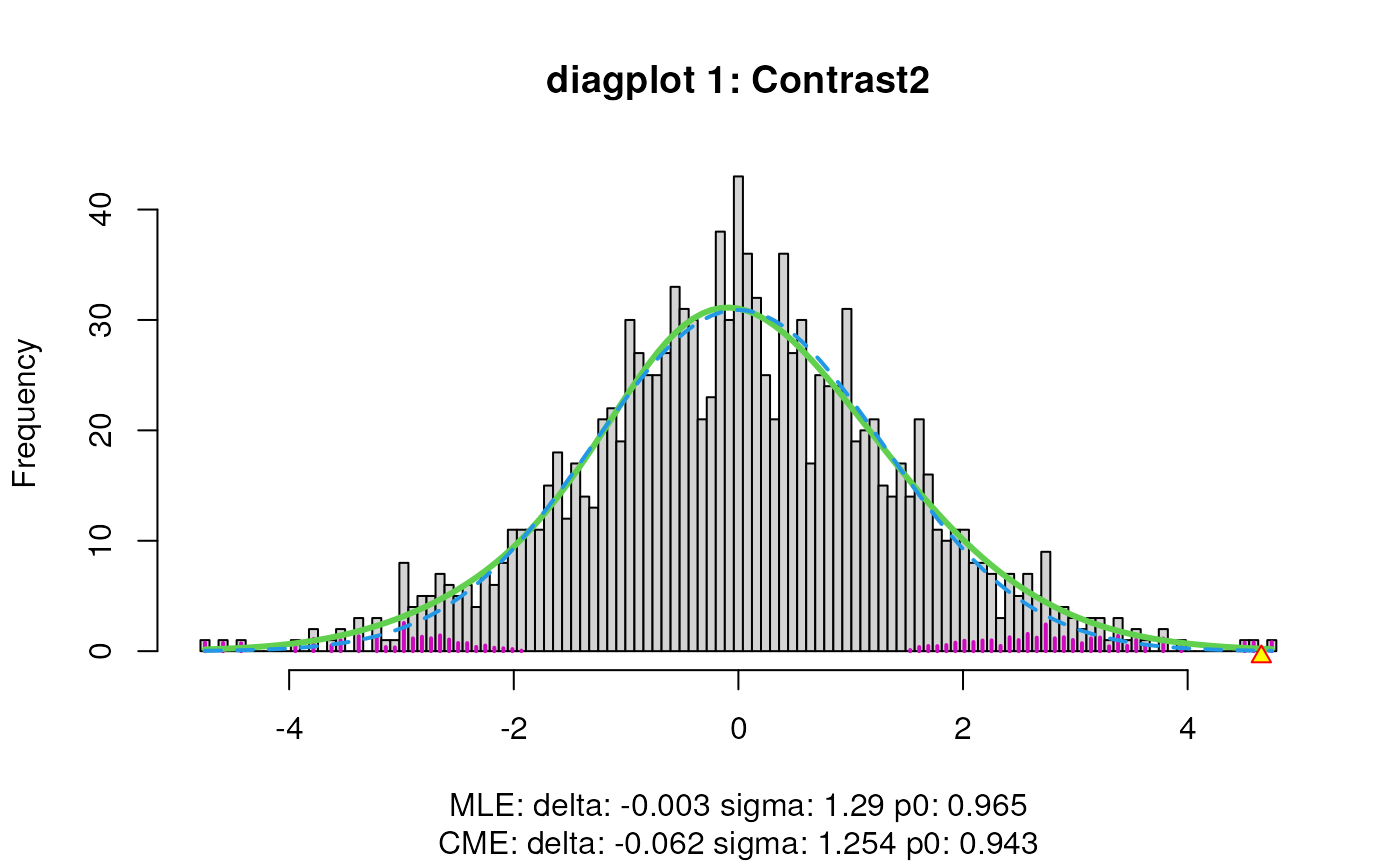
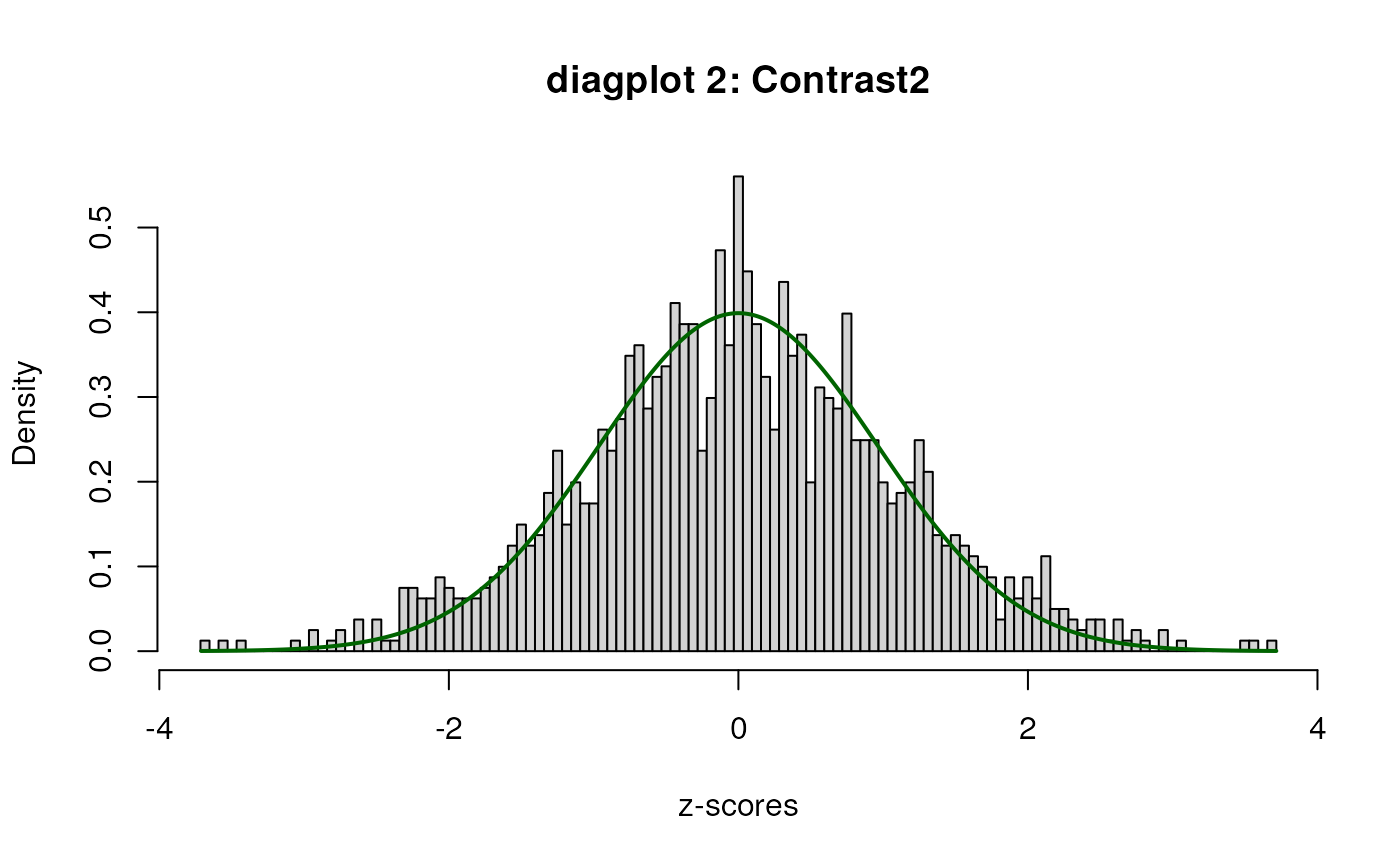
sumExp <- testDTU(object = sumExp,
contrasts = L,
diagplot1 = FALSE,
diagplot2 = FALSE,
sort = FALSE)
 group1 <- rownames(colData(sumExp))[colData(sumExp)$group ==
"VISp.L5_IT_VISp_Hsd11b1_Endou"]
group2 <- rownames(colData(sumExp))[colData(sumExp)$group ==
"ALM.L5_IT_ALM_Tnc"]
plots <- plotDTU(
object = sumExp,
contrast = "Contrast1",
groups = list(group1, group2),
coefficients = list(c(0, 0, 1), c(0, 1, 0)),
summaryStat = "model",
transcripts = c("ENSMUST00000165123",
"ENSMUST00000165721",
"ENSMUST00000005067"),
genes = NULL,
top.n = 6
)
group1 <- rownames(colData(sumExp))[colData(sumExp)$group ==
"VISp.L5_IT_VISp_Hsd11b1_Endou"]
group2 <- rownames(colData(sumExp))[colData(sumExp)$group ==
"ALM.L5_IT_ALM_Tnc"]
plots <- plotDTU(
object = sumExp,
contrast = "Contrast1",
groups = list(group1, group2),
coefficients = list(c(0, 0, 1), c(0, 1, 0)),
summaryStat = "model",
transcripts = c("ENSMUST00000165123",
"ENSMUST00000165721",
"ENSMUST00000005067"),
genes = NULL,
top.n = 6
)